

Genome-wide association study meta-analysis provides insights into the etiology of heart failure and its subtypes
- PMID: 40038546
- PMCID: PMC11985341
- DOI: 10.1038/s41588-024-02064-3
Genome-wide association study meta-analysis provides insights into the etiology of heart failure and its subtypes
Abstract
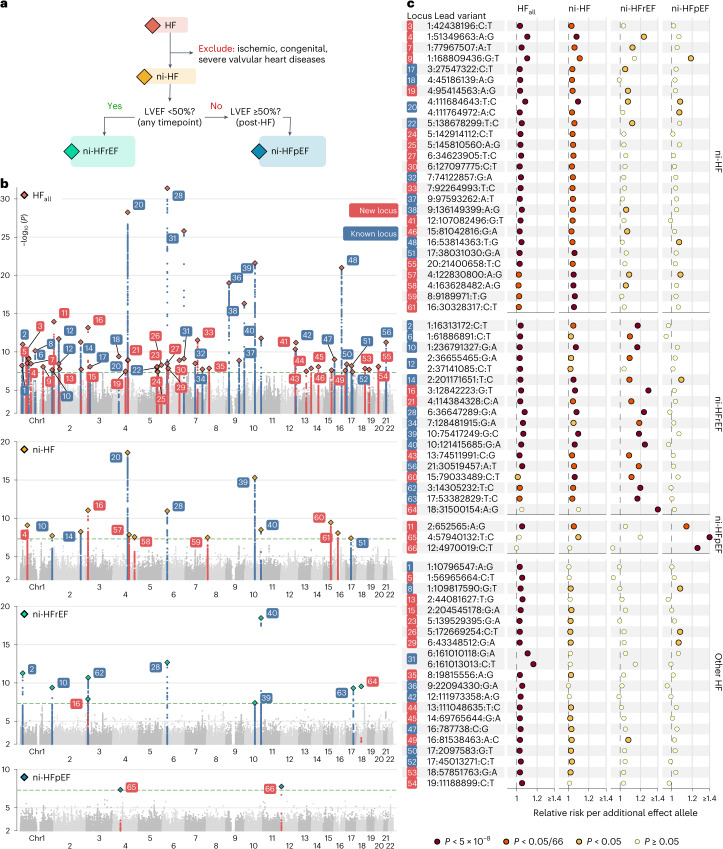
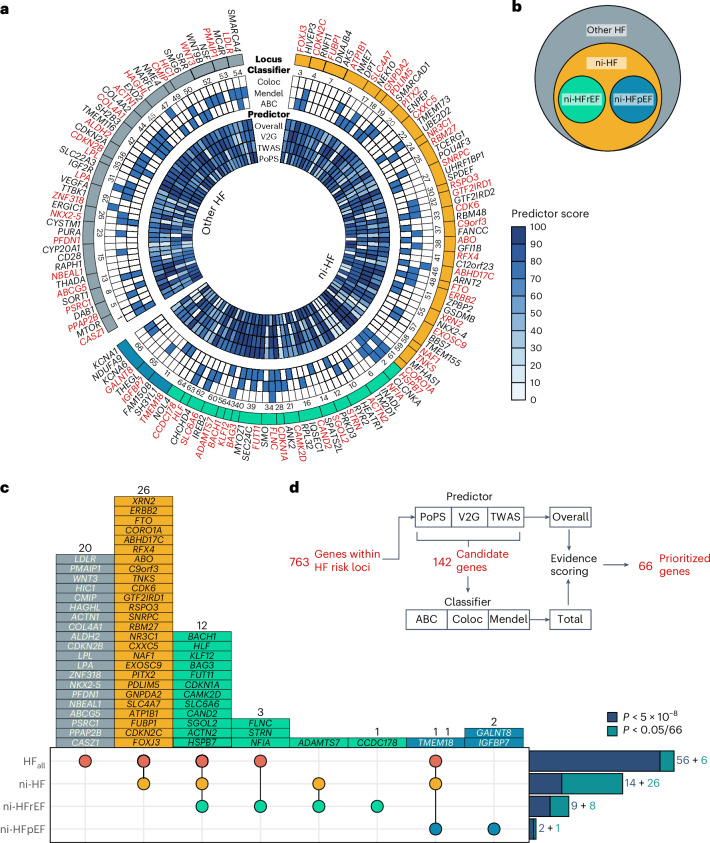
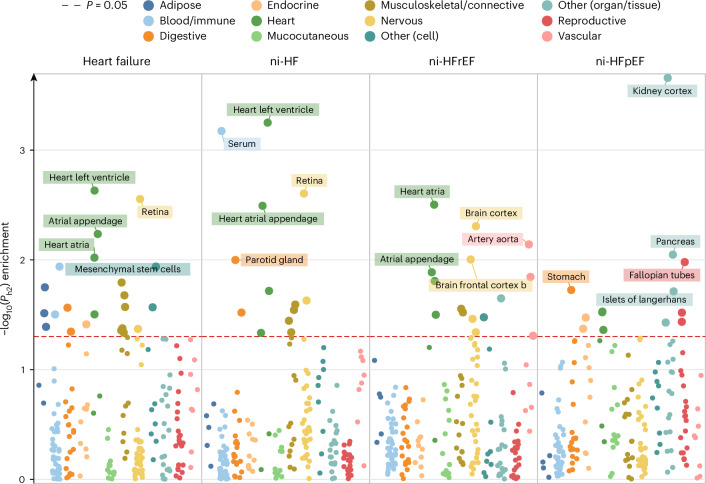
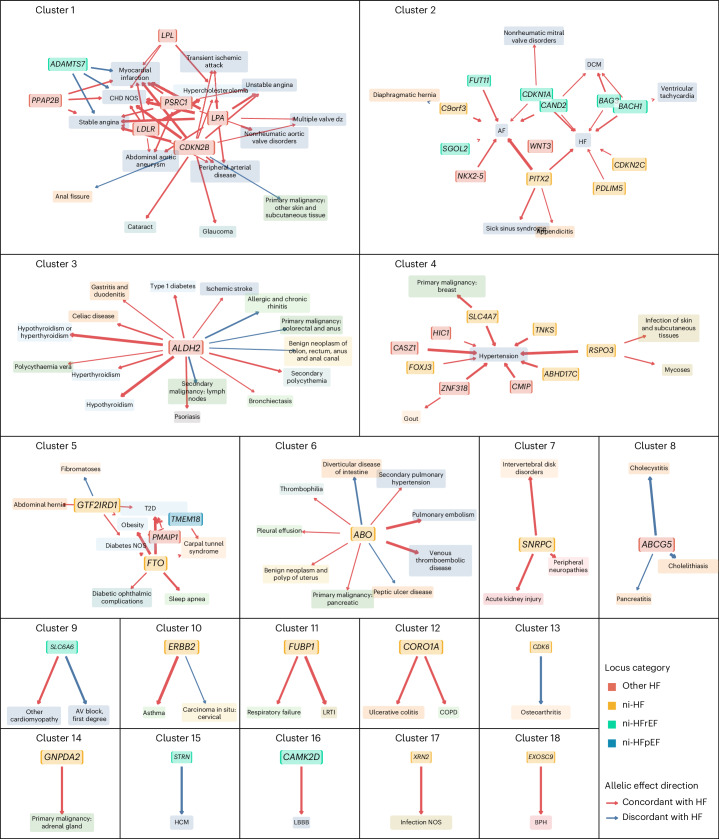
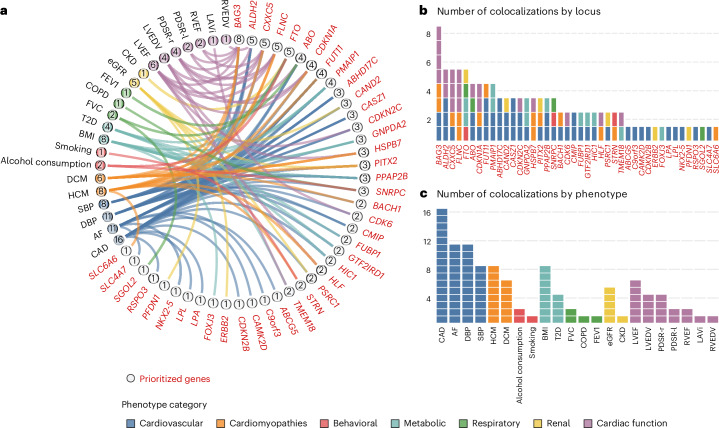
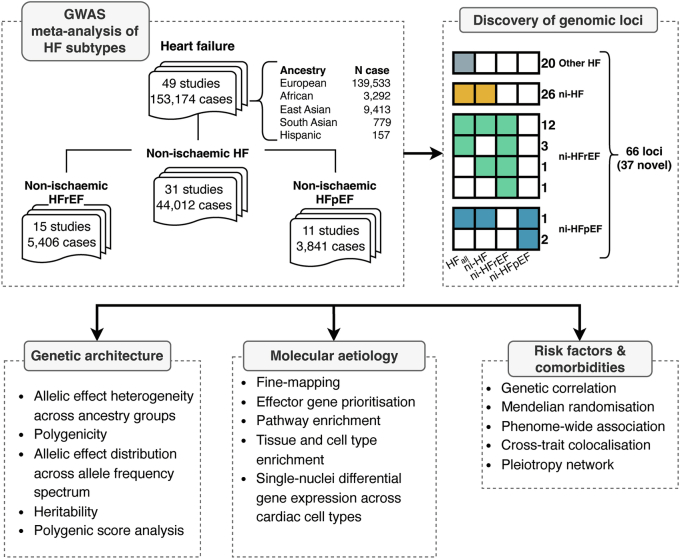
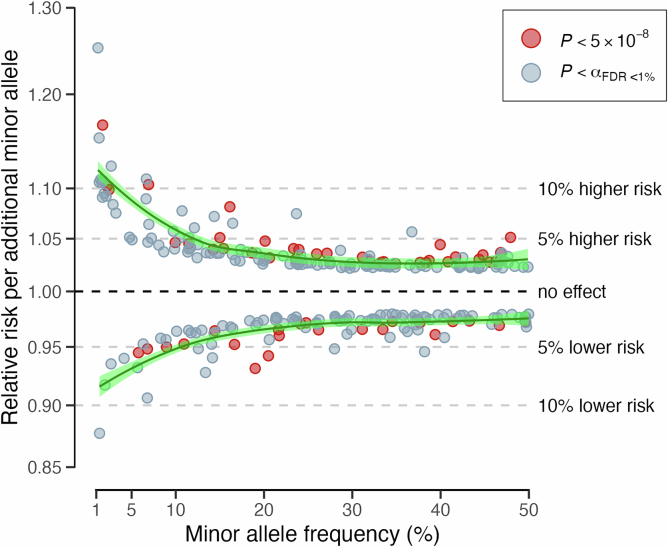
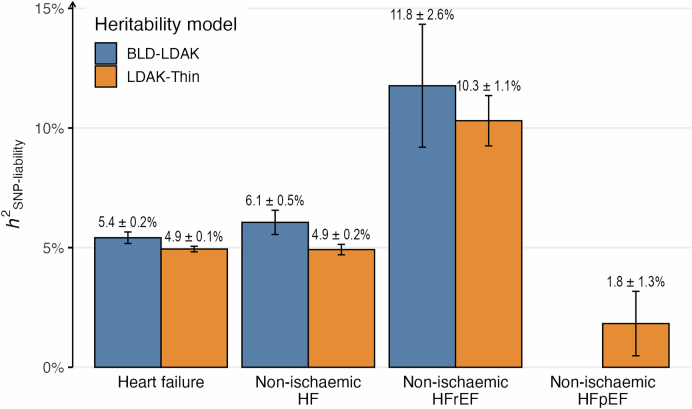
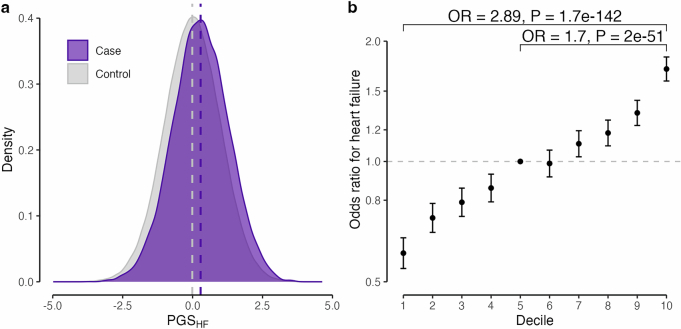
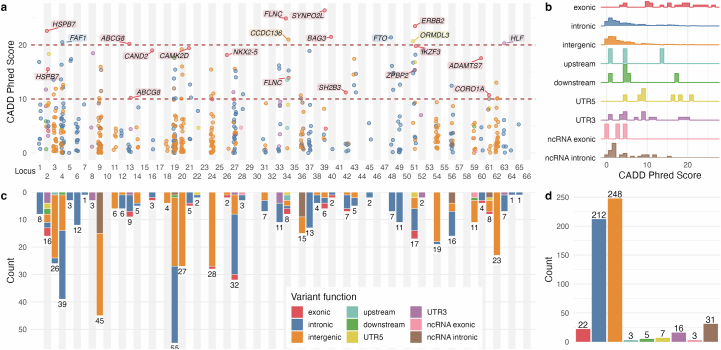
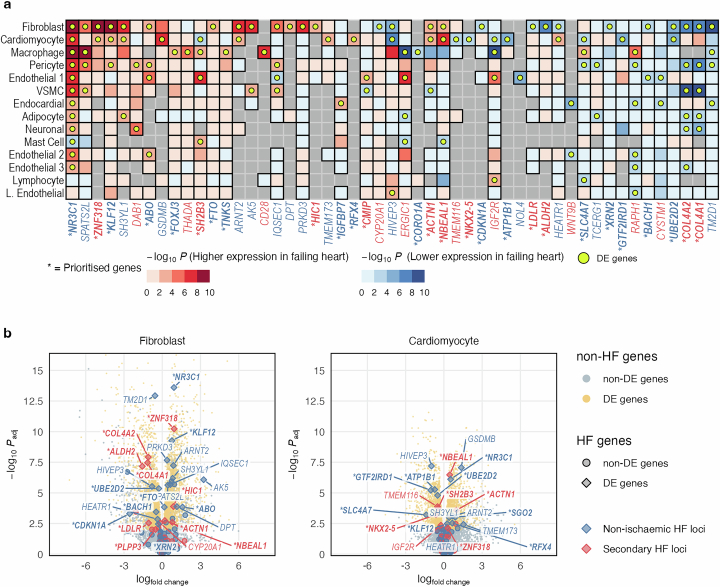
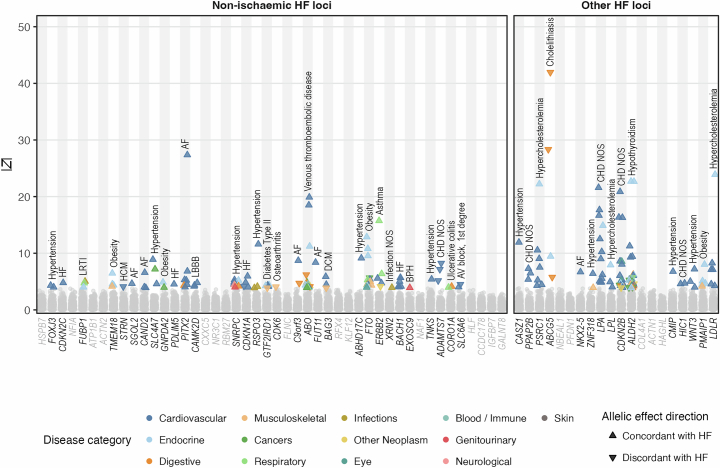
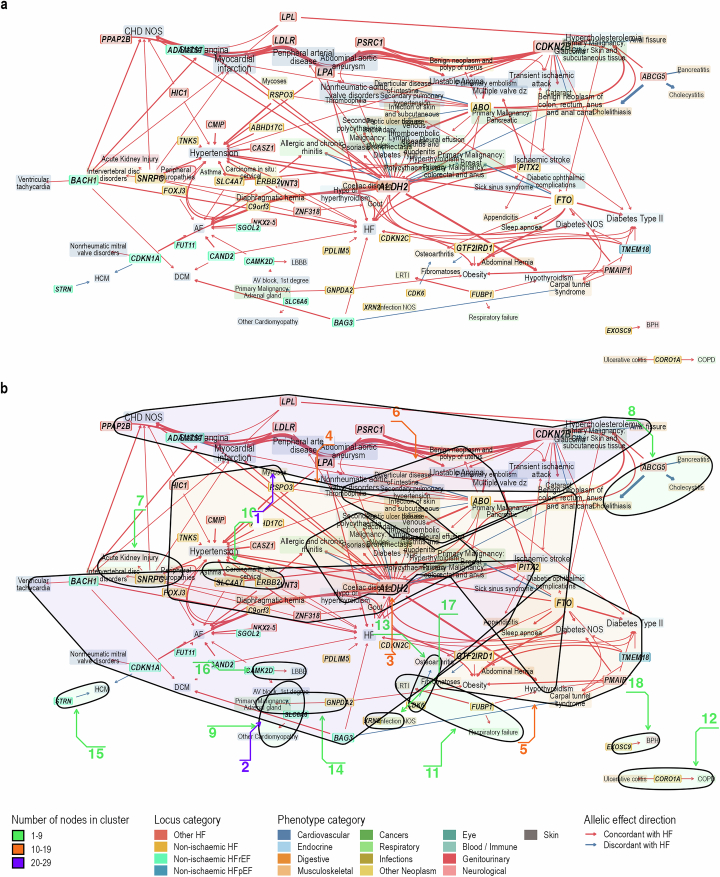
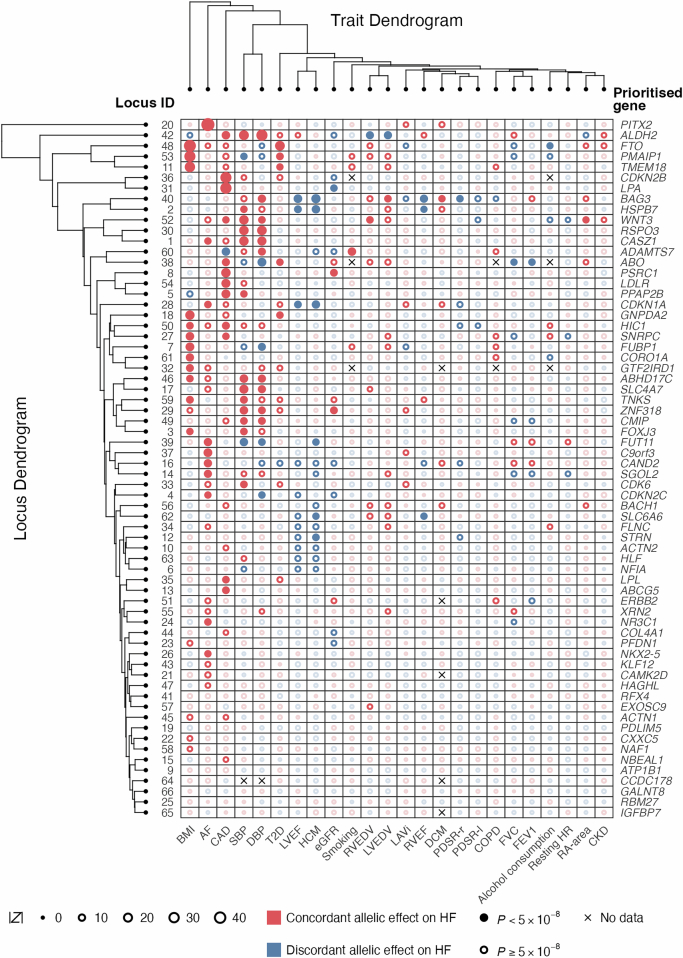
Heart failure (HF) is a major contributor to global morbidity and mortality. While distinct clinical subtypes, defined by etiology and left ventricular ejection fraction, are well recognized, their genetic determinants remain inadequately understood. In this study, we report a genome-wide association study of HF and its subtypes in a sample of 1.9 million individuals. A total of 153,174 individuals had HF, of whom 44,012 had a nonischemic etiology (ni-HF). A subset of patients with ni-HF were stratified based on left ventricular systolic function, where data were available, identifying 5,406 individuals with reduced ejection fraction and 3,841 with preserved ejection fraction. We identify 66 genetic loci associated with HF and its subtypes, 37 of which have not previously been reported. Using functionally informed gene prioritization methods, we predict effector genes for each identified locus, and map these to etiologic disease clusters through phenome-wide association analysis, network analysis and colocalization. Through heritability enrichment analysis, we highlight the role of extracardiac tissues in disease etiology. We then examine the differential associations of upstream risk factors with HF subtypes using Mendelian randomization. These findings extend our understanding of the mechanisms underlying HF etiology and may inform future approaches to prevention and treatment.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: R.T.L. received institutional research support from Pfizer. R.T.L. has served as a paid consultant for Health Lumen and FITFILE. J.S.W. has acted as a consultant for MyoKardia, Pfizer, Foresite Labs and Health Lumen and received institutional support from Bristol Myers Squibb and Pfizer. S.d.D. was supported through grants from AstraZeneca and Roche Molecular Science/DalCor. J.R.K. declares stock ownership in AbbVie, Abbott, Bristol Myers Squibb, Johnson & Johnson, Medtronic, Merck and Pfizer. N.A.M. received speaking honoraria from Amgen and is involved in clinical trials with Ionis, Amgen, Pfizer and Novartis. B.M.P. serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. C.T.R. received honoraria for scientific advisory boards and consulting from Anthos, Bayer, Bristol Myers Squibb, Daiichi Sankyo, Janssen and Pfizer and received institutional research grants from Anthos, AstraZeneca, Daiichi Sankyo, Janssen and Novartis. M.S.S. received substantial research grant support from Abbott Laboratories, Amgen, AstraZeneca, Bayer, Critical Diagnostics, Daiichi Sankyo, Eisai, Genzyme, Gilead, GlaxoSmithKline, Intarcia, Janssen Research and Development, The Medicines Company, MedImmune, Merck, Novartis, Poxel, Pfizer, Quark Pharmaceuticals, Roche Diagnostics and Takeda and has received consulting fees from Alnylam, AstraZeneca, Bristol Myers Squibb, CVS and Amgen. A.A.V. received consultancy fees and/or research support from AnaCardia, AstraZeneca, Bayer, BMS, Boehringer Ingelheim, Corteria, Cytokinetics, Eli Lilly, Moderna, Novartis, Novo Nordisk and Roche Diagnostics. M.-P.D. declares holding equity in Dalcor Pharmaceuticals, unrelated to this work. Members of the TIMI Study Group (ENGAGE, FOURIER, PEGASUS, SAVOR and SOLID) have received institutional research grant support through Brigham and Women’s Hospital from Abbott, Amgen, Anthos Therapeutics, ARCA Biopharma, AstraZeneca, Bayer HealthCare Pharmaceuticals, Daiichi Sankyo, Eisai, Intarcia, Ionis Pharmaceuticals, Janssen Research and Development, MedImmune, Merck, Novartis, Pfizer, Quark Pharmaceuticals, Regeneron Pharmaceuticals, Roche, Siemens Healthcare Diagnostics, Softcell Medical Limited, The Medicines Company, Zora Biosciences, Caremark, Dyrnamix, Esperon, IFM Pharmaceuticals and MyoKardia. The authors who are affiliated with deCODE genetics/Amgen and the authors affiliated with Pfizer declare competing financial interests as employees. The remaining authors declare no competing interests.
Figures


References
-
- Seferović, P. M. et al. Heart failure in cardiomyopathies: a position paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail.21, 553–576 (2019). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
