

SGV-caller: SARS-CoV-2 genome variation caller
- PMID: 40040978
- PMCID: PMC11876889
- DOI: 10.1016/j.heliyon.2025.e42613
SGV-caller: SARS-CoV-2 genome variation caller
Abstract
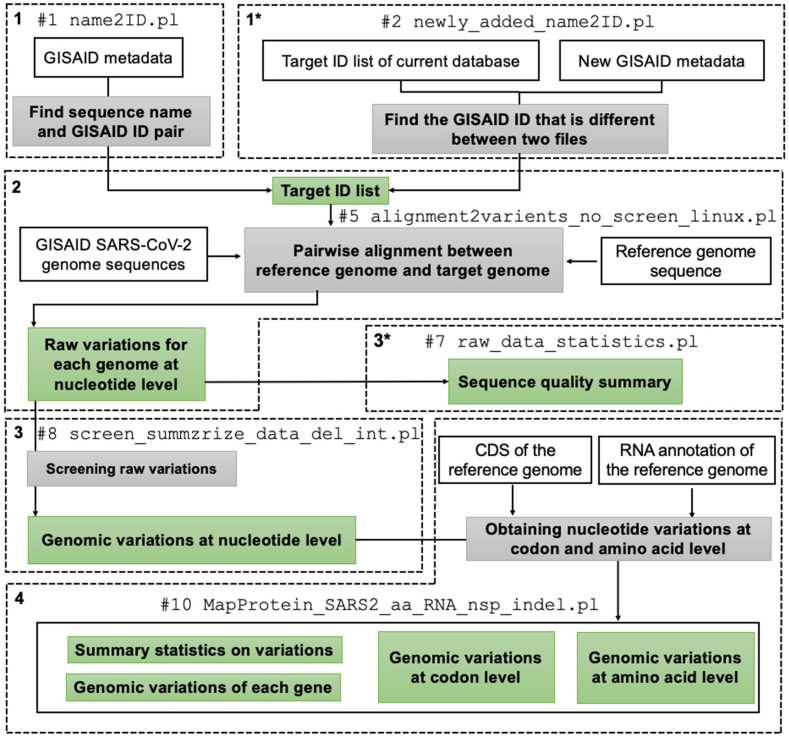
Given the pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), continuous analysis of its genomic variations at the nucleotide level is imperative to monitor the emergence of novel variants of concern. The Global Initiative on Sharing All Influenza Data (GISAID) serves as the de facto standard database for the genomic information of SARS-CoV-2. However, limitations of its data-sharing policy hinder the comprehensive analysis of genomic variations. To address this problem, we developed SGV-caller, a bioinformatics pipeline for analyzing the frequently updated GISAID database. SGV-caller compares input datasets with pre-existing databases and generates local databases encompassing nucleotide, amino acid, and codon-level genomic variations for each SARS-CoV-2 genome. Furthermore, SGV-caller accommodates SARS-CoV-2 genomes from non-GISAID sources as well as other viral genomes. SGV-caller source code and test data are available at https://github.com/wujiaqi06/SGV-caller.
Keywords: Bioinformatics pipeline; GISAID; Genome surveillance; Mutations; Viral genomes.
© 2025 The Authors.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
-
- Gangavarapu K., Latif A.A., Mullen J.L., Alkuzweny M., Hufbauer E., Tsueng G., et al. Outbreak.info genomic reports: scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. Nat. Methods. 2023;20(4):512–522. doi: 10.1038/s41592-023-01769-3. Epub 2023 Feb 23. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
