

A fast approach for structural and evolutionary analysis based on energetic profile protein comparison
- PMID: 40044697
- PMCID: PMC11882786
- DOI: 10.1038/s41467-025-57374-9
A fast approach for structural and evolutionary analysis based on energetic profile protein comparison
Erratum in
-
Author Correction: A fast approach for structural and evolutionary analysis based on energetic profile protein comparison.Nat Commun. 2025 Apr 4;16(1):3219. doi: 10.1038/s41467-025-58565-0. Nat Commun. 2025. PMID: 40185763 Free PMC article. No abstract available.
Abstract
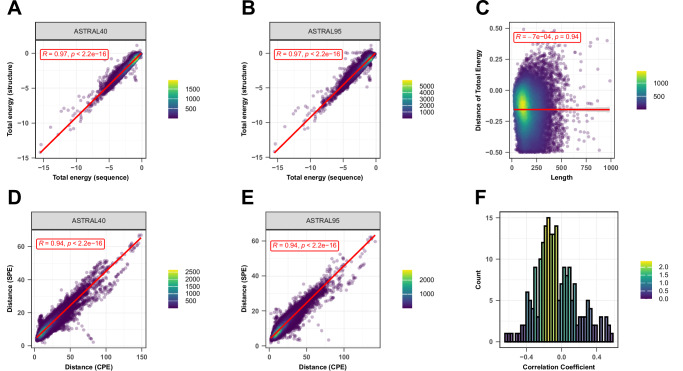
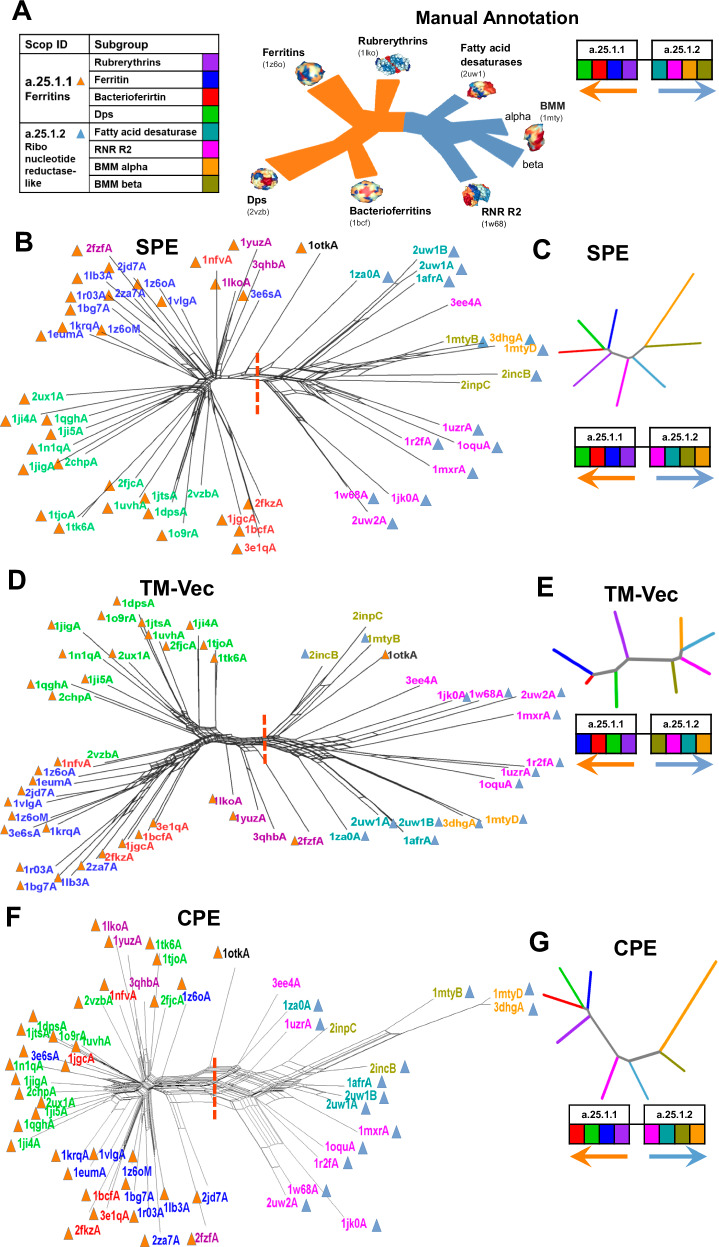
In structural bioinformatics, the efficiency of predicting protein similarity, function, and evolutionary relationships is crucial. Our approach proposed herein leverages protein energy profiles derived from a knowledge-based potential, deviating from traditional methods relying on structural alignment or atomic distances. This method assigns unique energy profiles to individual proteins, facilitating rapid comparative analysis for both structural similarities and evolutionary relationships across various hierarchical levels. Our study demonstrates that energy profiles contain substantial information about protein structure at class, fold, superfamily, and family levels. Notably, these profiles accurately distinguish proteins across species, illustrated by the classification of coronavirus spike glycoproteins and bacteriocin proteins. Introducing a separation measure based on energy profile similarity, our method shows significant correlation with a network-based approach, emphasizing the potential of energy profiles as efficient predictors for drug combinations with faster computational requirements. Our key insight is that the sequence-based energy profile strongly correlates with structure-derived energy, enabling rapid and efficient protein comparisons based solely on sequences.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures





References
-
- Du, Z. et al. pLM4ACE: A protein language model based predictor for antihypertensive peptide screening. Food Chem.431, 137162 (2024). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
