

Cross-species and mammal-to-mammal transmission of clade 2.3.4.4b highly pathogenic avian influenza A/H5N1 with PB2 adaptations
- PMID: 40044729
- PMCID: PMC11882949
- DOI: 10.1038/s41467-025-57338-z
Cross-species and mammal-to-mammal transmission of clade 2.3.4.4b highly pathogenic avian influenza A/H5N1 with PB2 adaptations
Abstract
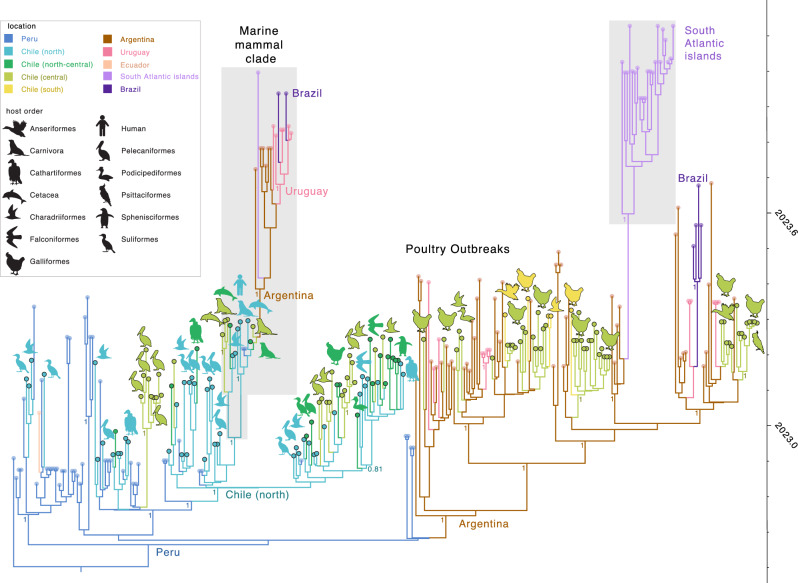
Highly pathogenic H5N1 avian influenza viruses (HPAIV) belonging to lineage 2.3.4.4b emerged in Chile in December 2022, leading to mass mortality events in wild birds, poultry, and marine mammals and one human case. We detected HPAIV in 7,33% (714/9745) of cases between December 2022-April 2023 and sequenced 177 H5N1 virus genomes from poultry, marine mammals, a human, and wild birds spanning >3800 km of Chilean coastline. Chilean viruses were closely related to Peru's H5N1 outbreak, consistent with north-to-south spread down the Pacific coastline. One human virus and nine marine mammal viruses in Chile had the rare PB2 D701N mammalian-adaptation mutation and clustered phylogenetically despite being sampled 5 weeks and hundreds of kilometers apart. These viruses shared additional genetic signatures, including another mammalian PB2 adaptation (Q591K, n = 6), synonymous mutations, and minor variants. Several mutations were detected months later in sealions in the Atlantic coast, indicating that the pinniped outbreaks on the west and east coasts of South America are genetically linked. These data support sustained mammal-to-mammal transmission of HPAIV in marine mammals over thousands of kilometers of Chile's Pacific coastline, which subsequently continued through the Atlantic coastline.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures






References
-
- Bessière, P., Dupré, G. & Volmer, R. Controlling the emergence of highly pathogenic avian influenza viruses: one of the challenges of the 21st century?. Virologie26, 343–354 (2022). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
