

TBK1 and IKKε prevent premature cell death by limiting the activity of both RIPK1 and NLRP3 death pathways
- PMID: 40053580
- PMCID: PMC11887814
- DOI: 10.1126/sciadv.adq1047
TBK1 and IKKε prevent premature cell death by limiting the activity of both RIPK1 and NLRP3 death pathways
Abstract
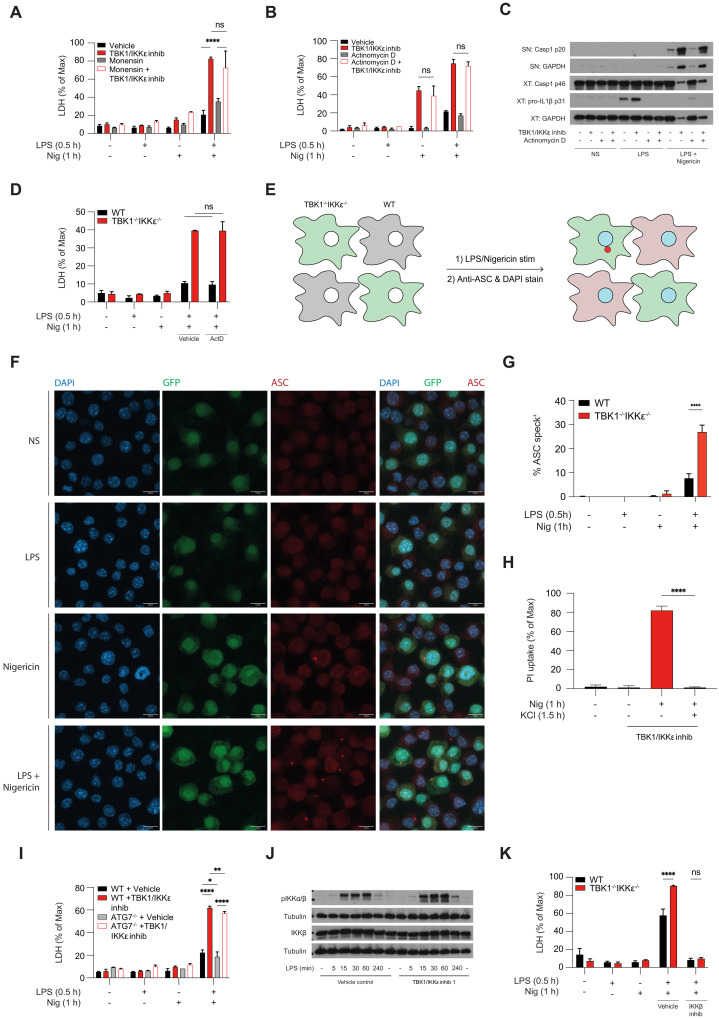
The loss of TBK1, or both TBK1 and the related kinase IKKε, results in uncontrolled cell death-driven inflammation. Here, we show that the pathway leading to cell death depends on the nature of the activating signal. Previous models suggest that in steady state, TBK1/IKKε-deficient cells die slowly and spontaneously predominantly by uncontrolled tumor necrosis factor-RIPK1-driven death. However, upon infection of cells that express the NLRP3 inflammasome, (e.g., macrophages), with pathogens that activate this pathway (e.g., Listeria monocytogenes), TBK1/IKKε-deficient cells die rapidly, prematurely, and exclusively by enhanced NLRP3-driven pyroptosis. Even infection with the RIPK1-activating pathogen, Yersinia pseudotuberculosis, results in enhanced RIPK1-caspase-8 activation and enhanced secondary NLRP3 activation. Mechanistically, TBK1/IKKε control endosomal traffic, and their loss disrupts endosomal homeostasis, thereby signaling cell stress. This results in premature NLRP3 activation even upon sensing "signal 2" alone, without the obligatory "signal 1." Collectively, TBK1/IKKε emerge as a central brake in limiting death-induced inflammation by both RIPK1 and NLRP3 death-inducing pathways.
Figures






References
-
- Balka K. R., Louis C., Saunders T. L., Smith A. M., Calleja D. J., D’Silva D. B., Moghaddas F., Tailler M., Lawlor K. E., Zhan Y., Burns C. J., Wicks I. P., Miner J. J., Kile B. T., Masters S. L., De Nardo D., TBK1 and IKKε act redundantly to mediate STING-induced NF-κB responses in myeloid cells. Cell Rep. 31, 107492 (2020). - PubMed
-
- Xu D., Jin T., Zhu H., Chen H., Ofengeim D., Zou C., Mifflin L., Pan L., Amin P., Li W., Shan B., Naito M. G., Meng H., Li Y., Pan H., Aron L., Adiconis X., Levin J. Z., Yankner B. A., Yuan J., TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell 174, 1477–1491.e19 (2018). - PMC - PubMed
-
- Lafont E., Draber P., Rieser E., Reichert M., Kupka S., de Miguel D., Draberova H., von Massenhausen A., Bhamra A., Henderson S., Wojdyla K., Chalk A., Surinova S., Linkermann A., Walczak H., TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol. 20, 1389–1399 (2018). - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
