

An atlas of RNA-dependent proteins in cell division reveals the riboregulation of mitotic protein-protein interactions
- PMID: 40057470
- PMCID: PMC11890761
- DOI: 10.1038/s41467-025-57671-3
An atlas of RNA-dependent proteins in cell division reveals the riboregulation of mitotic protein-protein interactions
Abstract
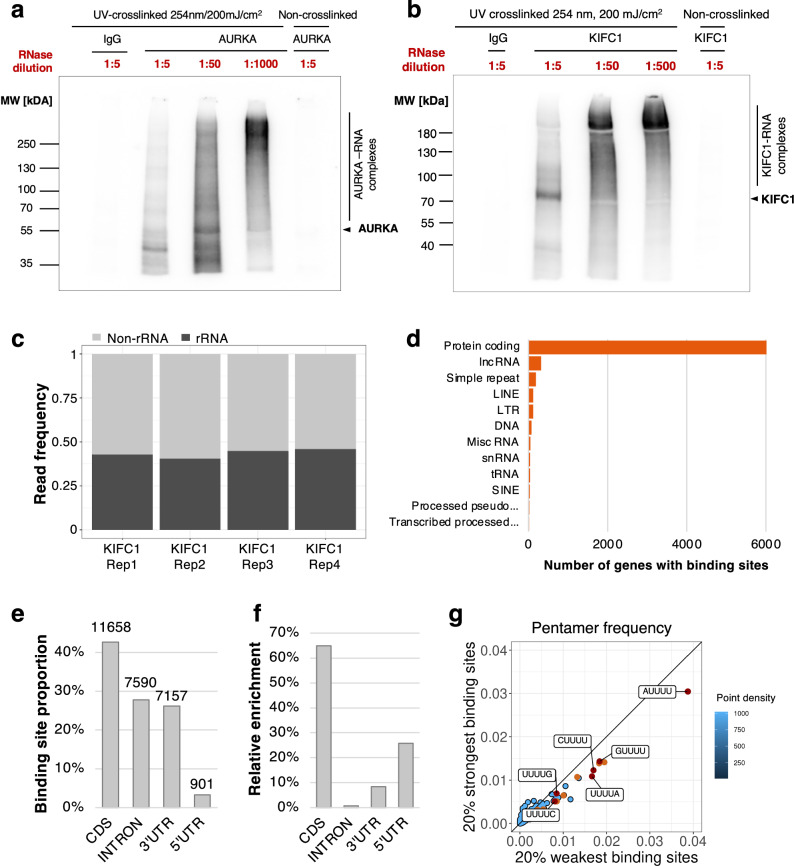
Ribonucleoprotein complexes are dynamic assemblies of RNA with RNA-binding proteins, which modulate the fate of RNA. Inversely, RNA riboregulates the interactions and functions of the associated proteins. Dysregulation of ribonucleoprotein functions is linked to diseases such as cancer and neurological disorders. In dividing cells, RNA and RNA-binding proteins are present in mitotic structures, but their impact on cell division remains unclear. By applying the proteome-wide R-DeeP strategy to cells synchronized in mitosis versus interphase integrated with the RBP2GO knowledge, we provided an atlas of RNA-dependent proteins in cell division, accessible at R-DeeP3.dkfz.de. We uncovered AURKA, KIFC1 and TPX2 as unconventional RNA-binding proteins. KIFC1 was identified as a new substrate of AURKA, and new TPX2-interacting protein. Their pair-wise interactions were RNA dependent. In addition, RNA stimulated AURKA kinase activity and stabilized its conformation. In this work, we highlighted riboregulation of major mitotic factors as an additional complexity level of cell division.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare the following competing interests: S.D. is co-owner of siTOOLs Biotech, Martinsried, Germany, without relation to this work. This study is part of the PhD thesis of V.R. The other authors declare no competing interests.
Figures







Update of
-
An atlas of RNA-dependent proteins in cell division reveals the riboregulation of mitotic protein-protein interactions.bioRxiv [Preprint]. 2024 Sep 26:2024.09.25.614981. doi: 10.1101/2024.09.25.614981. bioRxiv. 2024. Update in: Nat Commun. 2025 Mar 08;16(1):2325. doi: 10.1038/s41467-025-57671-3. PMID: 39386702 Free PMC article. Updated. Preprint.
References
-
- Dreyfuss, G., Kim, V. N. & Kataoka, N. Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol.3, 195–205 (2002). - PubMed
-
- Mitchell, S. F. & Parker, R. Principles and properties of eukaryotic mRNPs. Mol. Cell54, 547–558 (2014). - PubMed
-
- Gebauer, F., Schwarzl, T., Valcarcel, J. & Hentze, M. W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet.22, 185–198 (2021). - PubMed
-
- Mayr, C. Regulation by 3’-Untranslated Regions. Annu. Rev. Genet.51, 171–194 (2017). - PubMed
-
- Hentze, M. W., Castello, A., Schwarzl, T. & Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol.19, 327–341 (2018). - PubMed
MeSH terms
Substances
Grants and funding
- P30CA023108 DCC Core grant/Center for Strategic Scientific Initiatives, National Cancer Institute (NCI Center for Strategic Scientific Initiatives)
- BWST-ISF2019-027/Baden-Württemberg Stiftung (Baden-Württemberg Foundation)
- P30 CA023108/CA/NCI NIH HHS/United States
- DFG ZA 881/6-1 (project 541627224)/Deutsche Forschungsgemeinschaft (German Research Foundation)
- 70113919/Deutsche Krebshilfe (German Cancer Aid)
LinkOut - more resources
Full Text Sources
Miscellaneous
