

T-cell Dependency of Tumor Regressions and Complete Responses with RAS(ON) Multi-selective Inhibition in Preclinical Models of Pancreatic Ductal Adenocarcinoma
- PMID: 40057911
- PMCID: PMC12319402
- DOI: 10.1158/2159-8290.CD-24-1475
T-cell Dependency of Tumor Regressions and Complete Responses with RAS(ON) Multi-selective Inhibition in Preclinical Models of Pancreatic Ductal Adenocarcinoma
Abstract
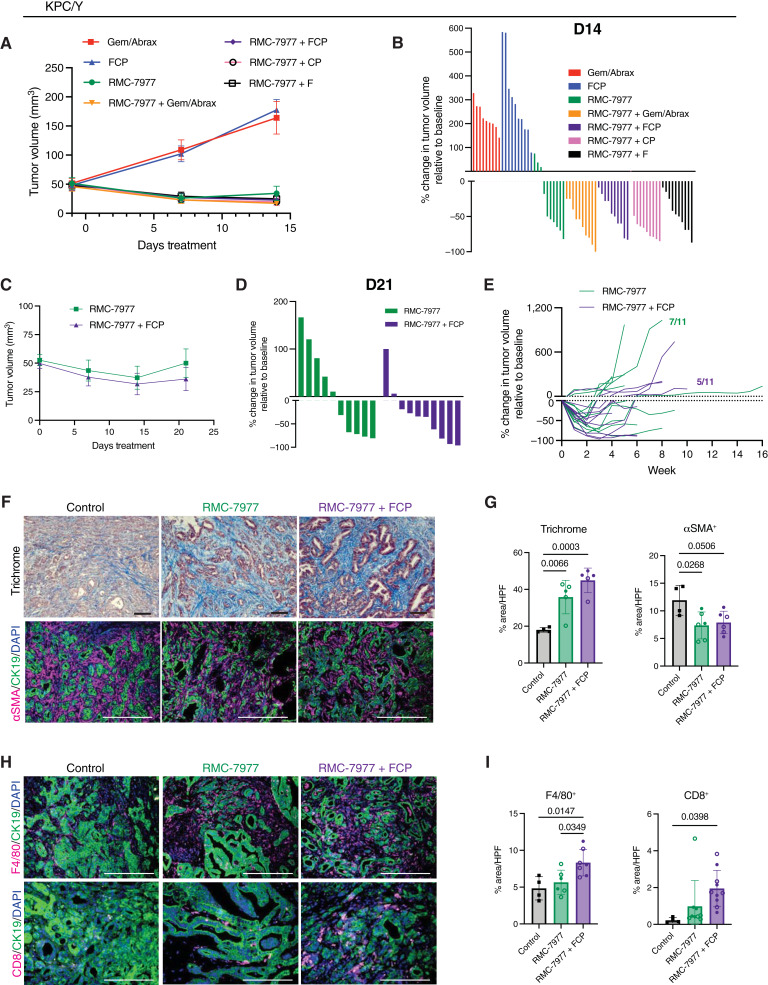
Activating mutations in KRAS drive tumorigenesis in pancreatic ductal adenocarcinoma (PDAC), promoting tumor cell proliferation and contributing to an immunosuppressive tumor microenvironment, rendering PDAC tumors insensitive to immunotherapy. RAS(ON) multi-selective inhibitors, such as daraxonrasib (RMC-6236) and RMC-7977, target the active state of RAS, with potent antitumor activity in PDAC murine models. In this study, we report that RAS(ON) multi-selective inhibition led to rapid and profound PDAC regressions in immunocompetent mice, decreasing myeloid cells and increasing T cells and macrophages in the tumor microenvironment. The depth and duration of tumor regression depended on T cells and conventional dendritic cells. Moreover, the combination of RAS(ON) multi-selective inhibitors with immunotherapy conferred deeper and more durable tumor regressions, including complete responses not seen with either treatment alone. In summary, concurrent inhibition of mutant and wild-type RAS is active in concert with T-cell immunotherapy, revealing RAS(ON) multi-selective inhibitors as a potential therapeutic immuno-sensitizing strategy in PDAC.
Significance: RAS(ON) multi-selective inhibitors enhance antitumor immunity in preclinical models of PDAC, resulting in deeper and more durable responses when combined with immunotherapy. These findings support the clinical evaluation of immune-based strategies that may prolong the response to RAS inhibitor therapies as well as overcome issues of therapeutic resistance to inhibitors alone. See related commentary by Lasse Opsahl and Pasca di Magliano, p. 1537.
©2025 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
N. Tovbis Shifrin reports a patent for WO 2024/229406 pending. M. Singh reports a patent for WO 2022/060836 pending and a patent for WO 2024/229406 pending. E. Quintana reports a patent for WO 2024/229406 pending. M. Menard reports a patent for WO 2024/229406 pending. R.H. Vonderheide reports other support from Revolution Medicines and grants from the NCI during the conduct of the study, as well as personal fees from Crossbow Therapeutics, Bristol Myers Squibb, EMD Sereno, and Grey Wolf Therapeutics outside the submitted work; in addition, R.H. Vonderheide has a patent for “Tumor antigens” issued, a patent for “Kras” epitopes issued, and a patent for “Cell therapy” issued, licensed, and with royalties paid from Therapeutics. B.Z. Stanger reports grants from Revolution Medicines during the conduct of the study and grants from Boehringer Ingelheim and personal fees from iTeos Therapeutics outside the submitted work. No disclosures were reported by the other authors.
Figures






References
-
- Carvalho PD, Guimarães CF, Cardoso AP, Mendonça S, Costa ÂM, Oliveira MJ, et al. KRAS oncogenic signaling extends beyond cancer cells to orchestrate the microenvironment. Cancer Res 2017;78:7–14. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
