

Analysis of Limited Proteolysis-Coupled Mass Spectrometry Data
- PMID: 40058498
- PMCID: PMC12036054
- DOI: 10.1016/j.mcpro.2025.100934
Analysis of Limited Proteolysis-Coupled Mass Spectrometry Data
Abstract
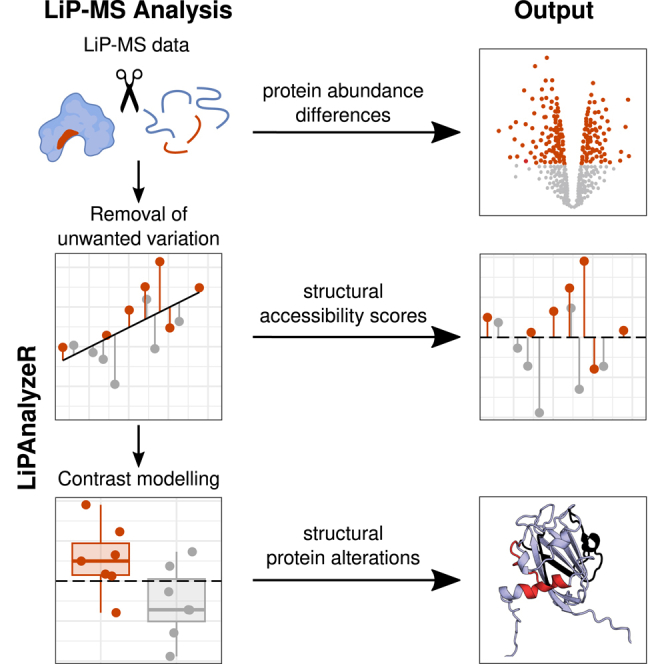
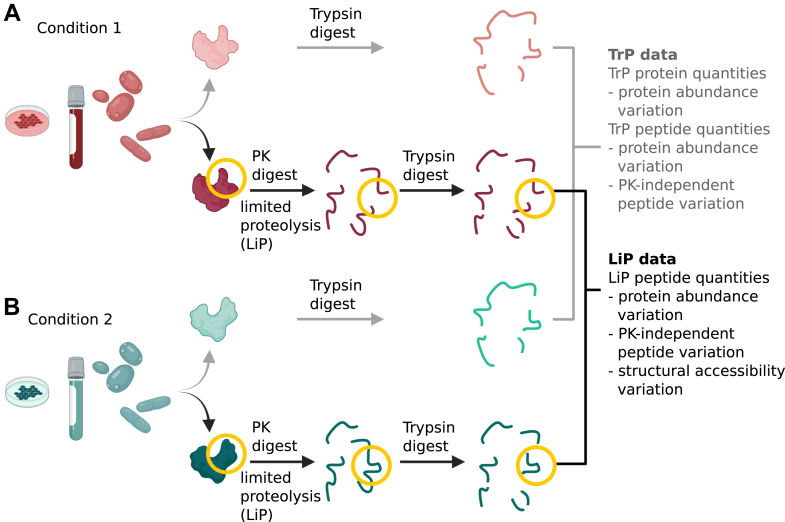
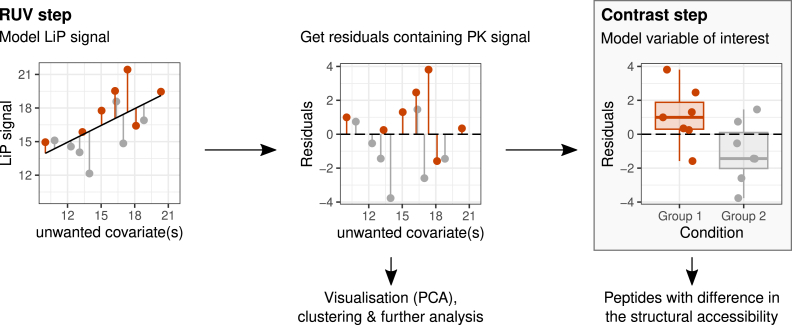
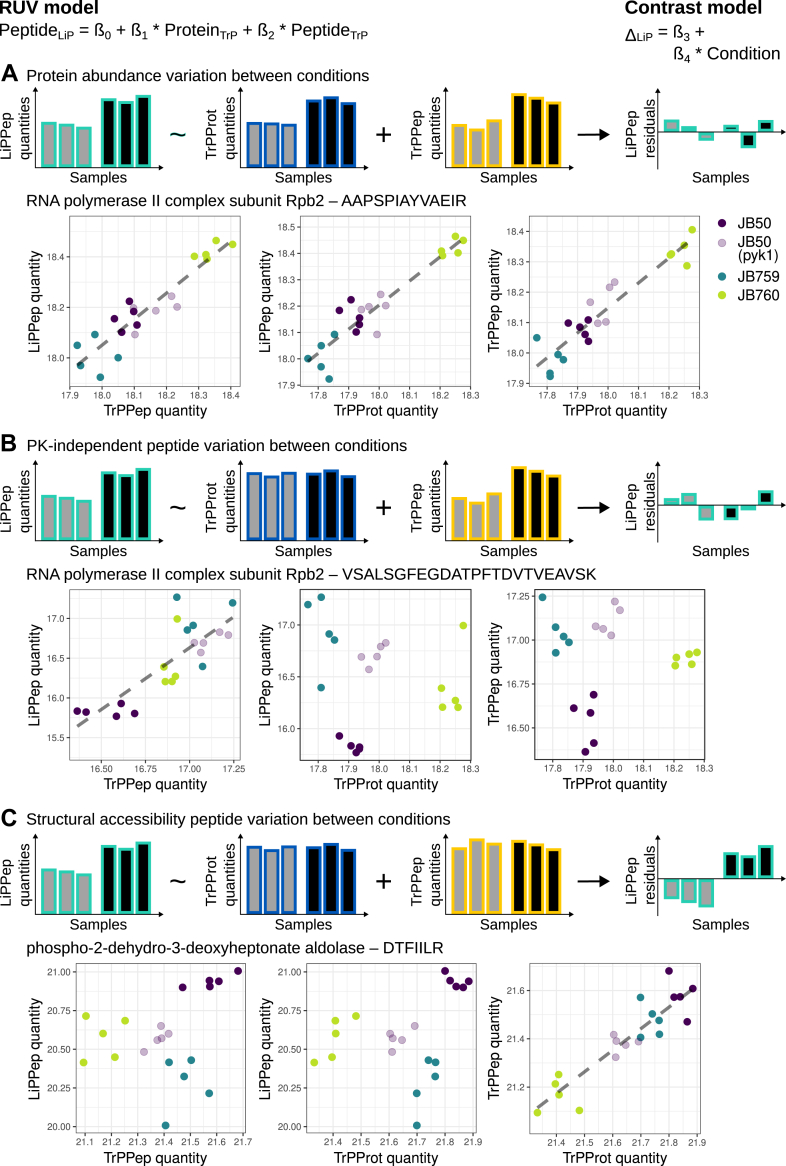
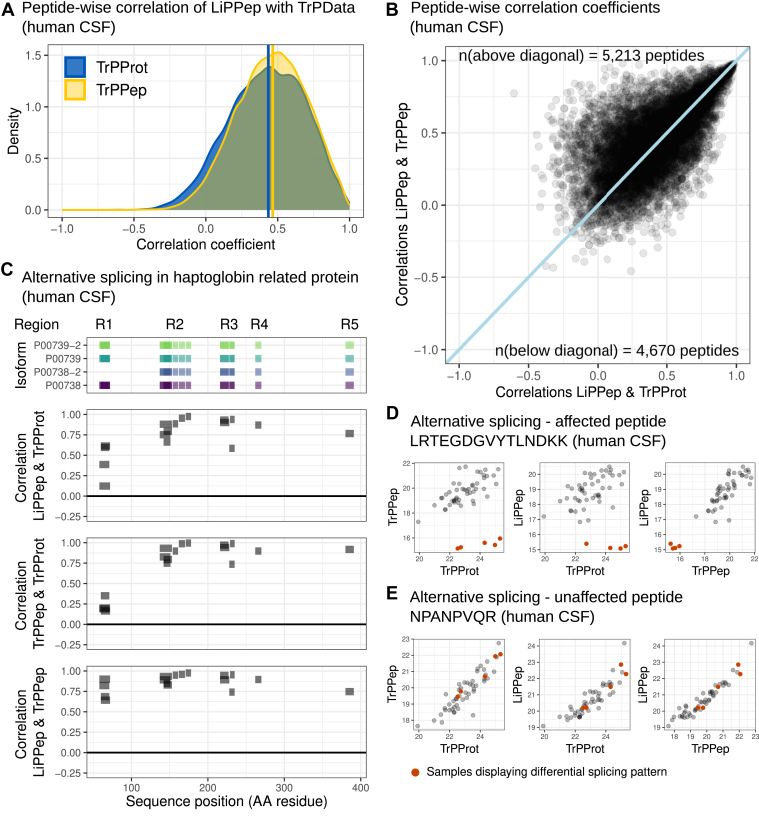
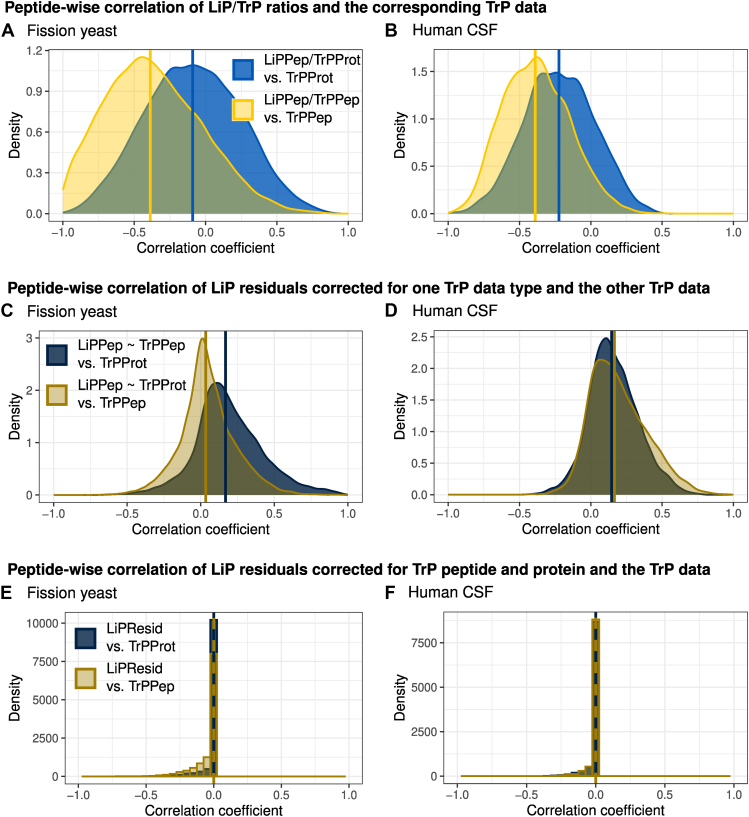
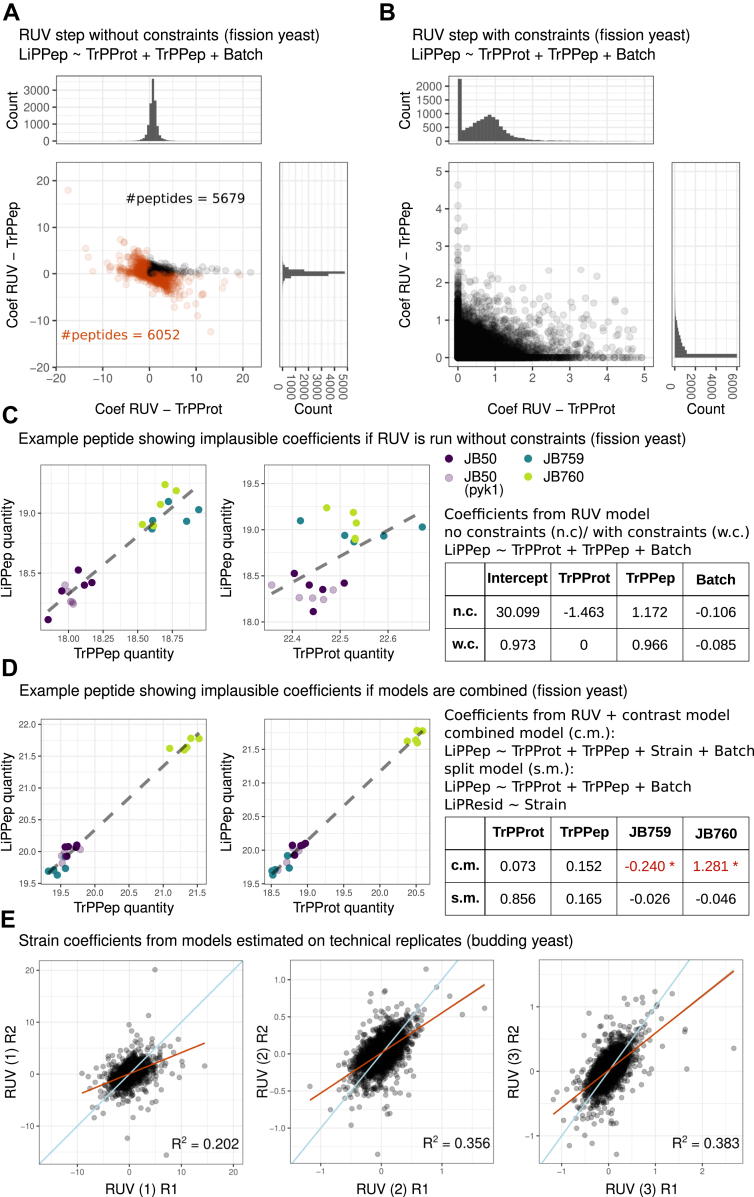
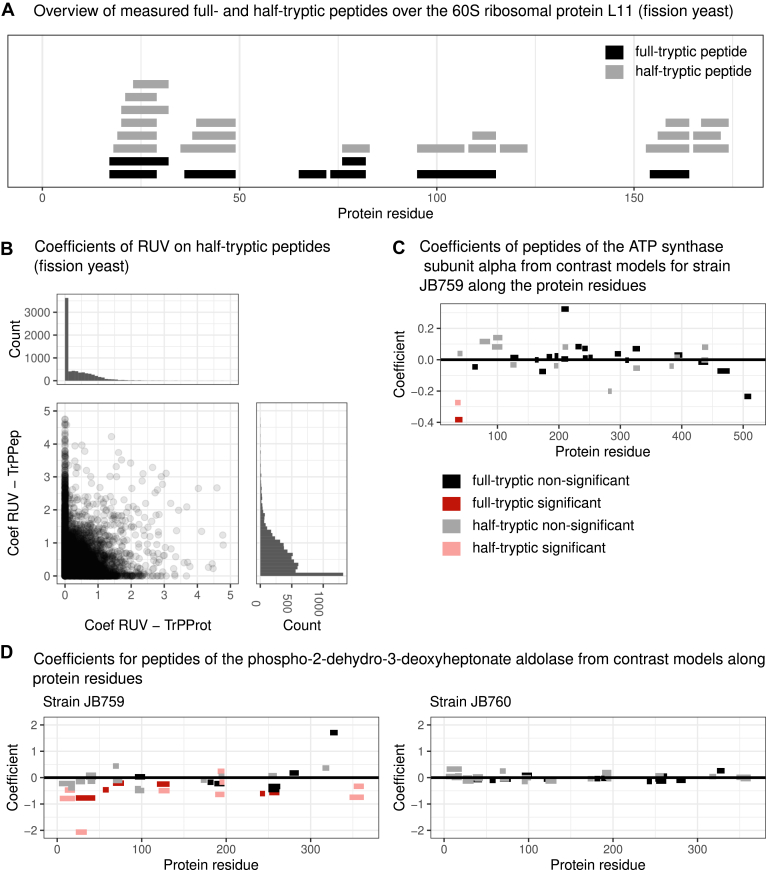
Limited proteolysis combined with mass spectrometry (LiP-MS) facilitates probing structural changes on a proteome-wide scale. This method leverages differences in the proteinase K accessibility of native protein structures to concurrently assess structural alterations for thousands of proteins in situ. Distinguishing different contributions to the LiP-MS signal, such as changes in protein abundance or chemical modifications, from structural protein alterations remains challenging. Here, we present the first comprehensive computational pipeline to infer structural alterations for LiP-MS data using a two-step approach. 1) We remove unwanted variations from the LiP signal that are not caused by protein structural effects and 2) infer the effects of variables of interest on the remaining signal. Using LiP-MS data from three species, we demonstrate that this approach outperforms previously employed approaches. Our framework provides a uniquely powerful approach for deconvolving LiP-MS signals and separating protein structural changes from changes in protein abundance, posttranslational modifications, and alternative splicing. Our approach may also be applied to analyze other types of peptide-centric structural proteomics data, such as FPOP or molecular painting data.
Keywords: MS data analysis; R package; limited proteolysis-coupled mass spectrometry (LiP-MS); protein structure; proteomics data analysis; statistical modelling.
Copyright © 2025 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest P. P. is a scientific advisor for the company Biognosys AG (Zurich, Switzerland) and an inventor of a patent licensed by Biognosys AG that covers the LiP-MS method used in this protocol. The remaining authors declare no competing interests.
Figures
Similar articles
-
Proteome-wide structural changes measured with limited proteolysis-mass spectrometry: an advanced protocol for high-throughput applications.Nat Protoc. 2023 Mar;18(3):659-682. doi: 10.1038/s41596-022-00771-x. Epub 2022 Dec 16. Nat Protoc. 2023. PMID: 36526727 Review.
-
Limited Proteolysis-Mass Spectrometry to Identify Metabolite-Protein Interactions.Methods Mol Biol. 2023;2554:69-89. doi: 10.1007/978-1-0716-2624-5_6. Methods Mol Biol. 2023. PMID: 36178621
-
Measuring protein structural changes on a proteome-wide scale using limited proteolysis-coupled mass spectrometry.Nat Protoc. 2017 Nov;12(11):2391-2410. doi: 10.1038/nprot.2017.100. Epub 2017 Oct 26. Nat Protoc. 2017. PMID: 29072706
-
Benchmarking of Quantitative Proteomics Workflows for Limited Proteolysis Mass Spectrometry.Mol Cell Proteomics. 2025 Apr;24(4):100945. doi: 10.1016/j.mcpro.2025.100945. Epub 2025 Mar 13. Mol Cell Proteomics. 2025. PMID: 40089063 Free PMC article.
-
Mass spectrometry analysis of the structural proteome.Curr Opin Struct Biol. 2020 Feb;60:57-65. doi: 10.1016/j.sbi.2019.10.006. Epub 2019 Dec 13. Curr Opin Struct Biol. 2020. PMID: 31841731 Review.
Cited by
-
Chaperone Dependency during Primary Protein Biogenesis Does Not Correlate with Chaperone Dependency during in vitro Refolding.bioRxiv [Preprint]. 2025 Jun 19:2025.06.16.659923. doi: 10.1101/2025.06.16.659923. bioRxiv. 2025. PMID: 40611891 Free PMC article. Preprint.
References
-
- Henzler-Wildman K., Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. - PubMed
-
- Tzeng S.-R., Kalodimos C.G. Protein activity regulation by conformational entropy. Nature. 2012;488:236–240. - PubMed
-
- Feng Y., De Franceschi G., Kahraman A., Soste M., Melnik A., Boersema P.J., et al. Global analysis of protein structural changes in complex proteomes. Nat. Biotechnol. 2014;32:1036–1044. - PubMed
-
- Savitski M.M., Reinhard F.B.M., Franken H., Werner T., Savitski M.F., Eberhard D., et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
