

Determining population structure from k-mer frequencies
- PMID: 40061228
- PMCID: PMC11890038
- DOI: 10.7717/peerj.18939
Determining population structure from k-mer frequencies
Abstract
Background: Understanding population structure within species provides information on connections among different populations and how they evolve over time. This knowledge is important for studies ranging from evolutionary biology to large-scale variant-trait association studies. Current approaches to determining population structure include model-based approaches, statistical approaches, and distance-based ancestry inference approaches.
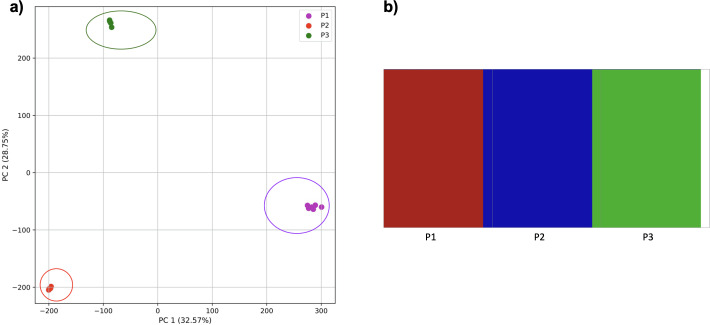
Methods: In this work, we identify population structure from DNA sequence data using an alignment-free approach. We use the frequencies of short DNA substrings from across the genome (k-mers) with principal component analysis (PCA). K-mer frequencies can be viewed as a summary statistic of a genome and have the advantage of being easily derived from a genome by counting the number of times a k-mer occurred in a sequence. In contrast, most population structure work employing PCA uses multi-locus genotype data (SNPs, microsatellites, or haplotypes). No genetic assumptions must be met to generate k-mers, whereas current population structure approaches often depend on several genetic assumptions and can require careful selection of ancestry informative markers to identify populations. We compare our k-mer based approach to population structure estimated using SNPs with both empirical and simulated data.
Results: In this work, we show that PCA is able to determine population structure just from the frequency of k-mers found in the genome. The application of PCA and a clustering algorithm to k-mer profiles of genomes provides an easy approach to detecting the number and composition of populations (clusters) present in the dataset. Using simulations, we show that results are at least comparable to population structure estimates using SNPs. When using human genomes from populations identified by the 1000 Genomes Project, the results are better than population structure estimates using SNPs from the same samples, and comparable to those found by a model-based approach using genetic markers from larger numbers of samples.
Conclusions: This study shows that PCA, together with the clustering algorithm, is able to detect population structure from k-mer frequencies and can separate samples of admixed and non-admixed origin. Using k-mer frequencies to determine population structure has the potential to avoid some challenges of existing methods and may even improve on estimates from small samples.
Keywords: Population differentiation; Population stratification; Population structure; k-mer frequencies; k-mers.
©2025 Hrytsenko et al.
Conflict of interest statement
The authors declare there are no competing interests.
Figures














References
-
- Andam CP, Challagundla L, Azarian T, Hanage WP, Robinson DA. Genetics and evolution of infectious diseases. Elsevier; 2017. Population structure of pathogenic bacteria; pp. 51–70. - DOI
-
- Andrews CA. Natural selection, genetic drift, and gene flow do not act in isolation in natural populations. Nature Education Knowledge. 2010;3:5.
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
