

Suppression of stress granule formation is a vulnerability imposed by mutant p53
- PMID: 40064891
- PMCID: PMC11894096
- DOI: 10.1038/s41467-025-57539-6
Suppression of stress granule formation is a vulnerability imposed by mutant p53
Abstract
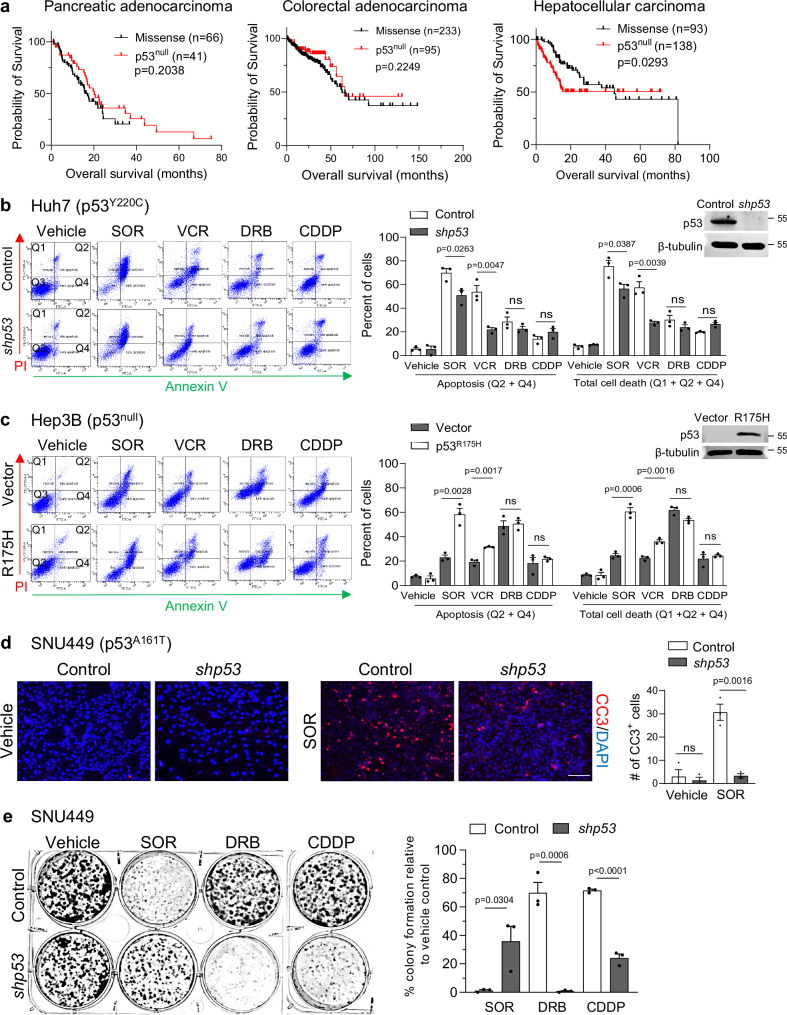
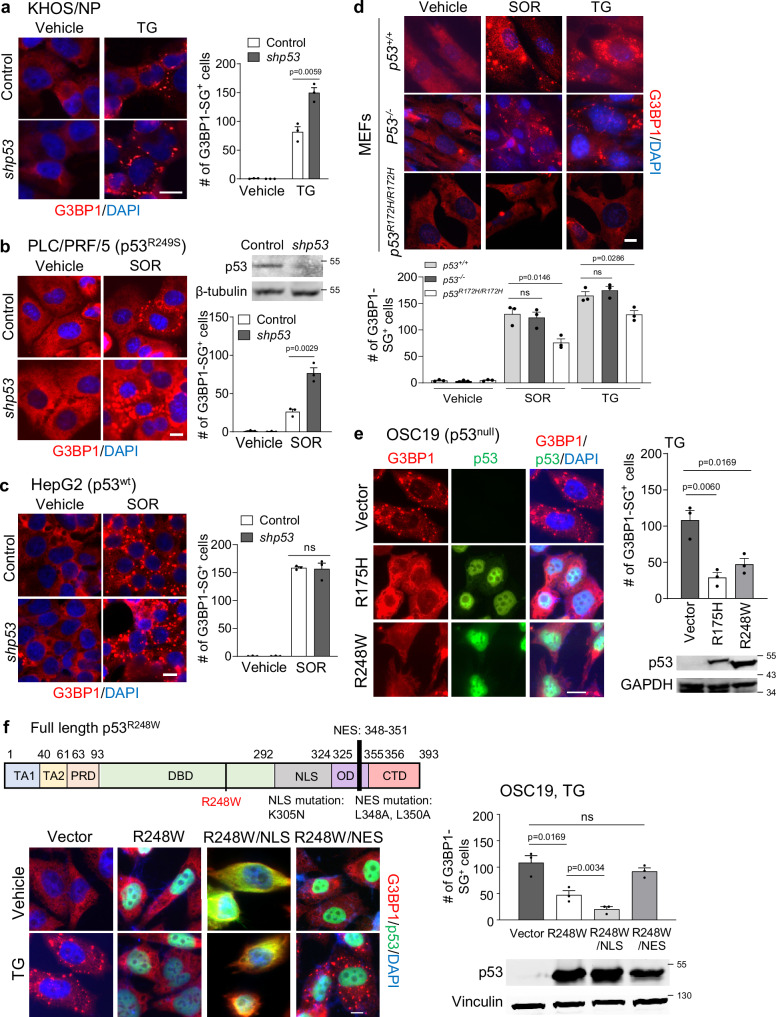
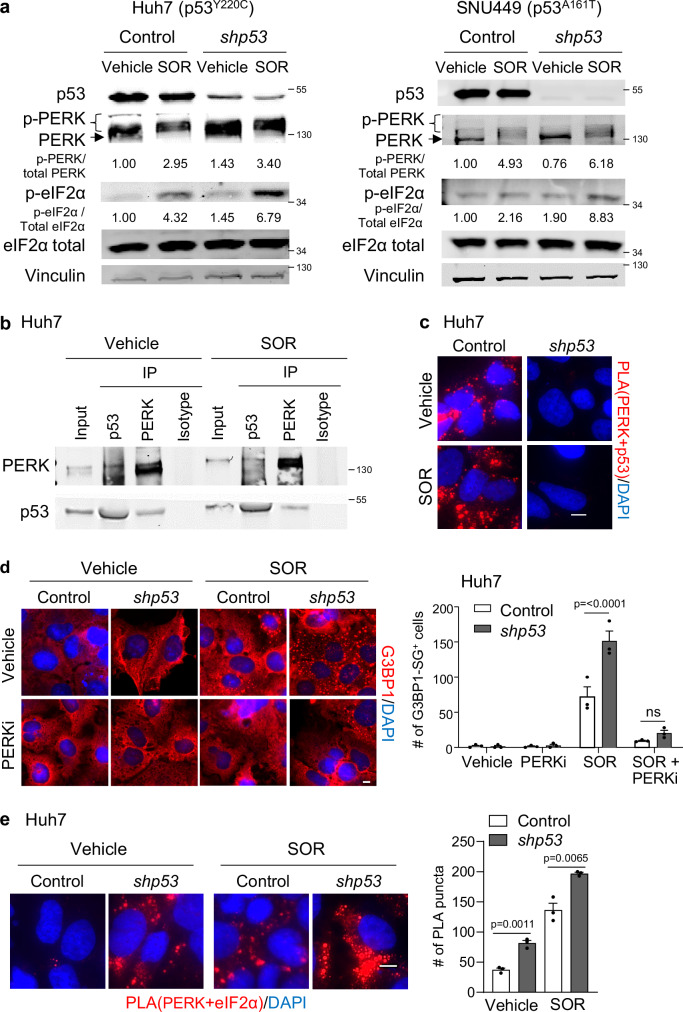
Missense mutations in the TP53 (p53) gene have been linked to malignant progression. However, our in-silico analyses reveal that hepatocellular carcinoma (HCC) patients with mutant p53 (mutp53) have better overall survival compared to those with p53-null (p53null) HCC, unlike other cancer types. Given the historical use of sorafenib (SOR) monotherapy for advanced HCC, we hypothesize that mutp53 increases sensitivity to SOR, a multikinase inhibitor that induces endoplasmic reticulum (ER) stress. Here we show that mutp53 inhibits stress granule (SG) formation by binding to an ER stress sensor, PKR-like ER kinase (PERK), and a key SG component, GAP SH3 domain-binding protein 1 (G3BP1), contributing to increased sensitivity of SG-competent cells and xenografts to ER stress inducers including SOR. Our study identifies a unique vulnerability imposed by mutp53, suggesting mutp53 as a biomarker for ER stress-inducing agents and highlighting the importance of SG inhibition for cancer treatment.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures





References
-
- Arimoto, K., Fukuda, H., Imajoh-Ohmi, S., Saito, H. & Takekawa, M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell Biol.10, 1324–1332 (2008). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
