

BET degraders reveal BRD4 disruption of 7SK and P-TEFb is critical for effective reactivation of latent HIV in CD4+ T-cells
- PMID: 40067013
- PMCID: PMC11998493
- DOI: 10.1128/jvi.01777-24
BET degraders reveal BRD4 disruption of 7SK and P-TEFb is critical for effective reactivation of latent HIV in CD4+ T-cells
Abstract
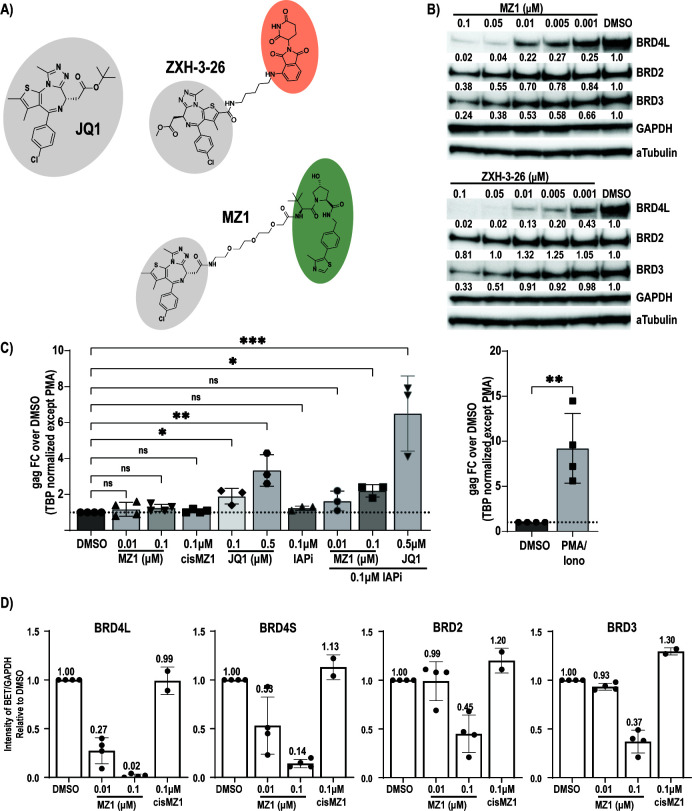
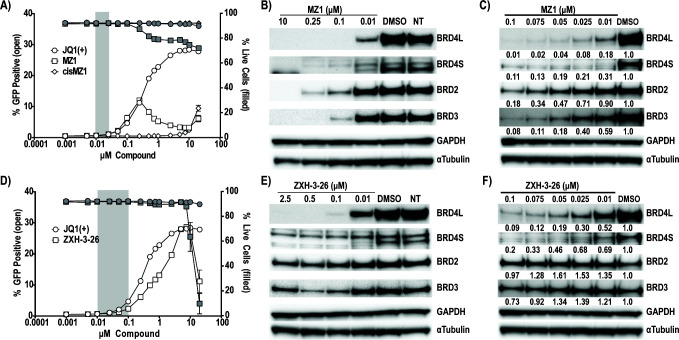
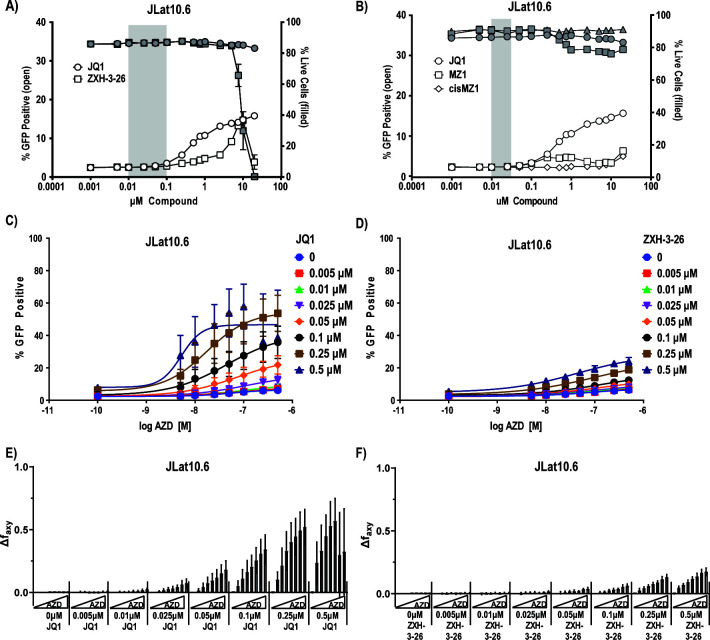
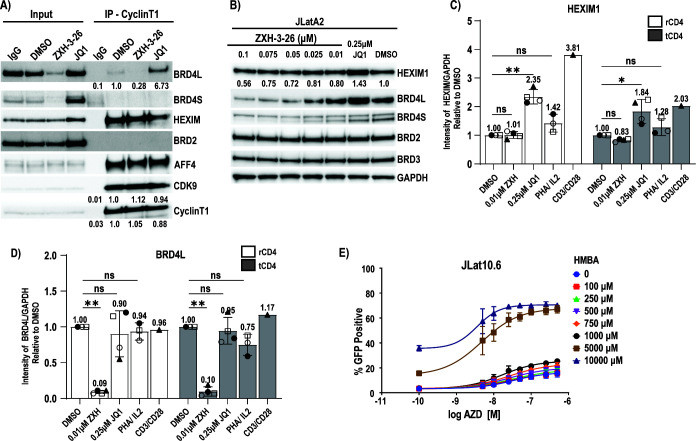
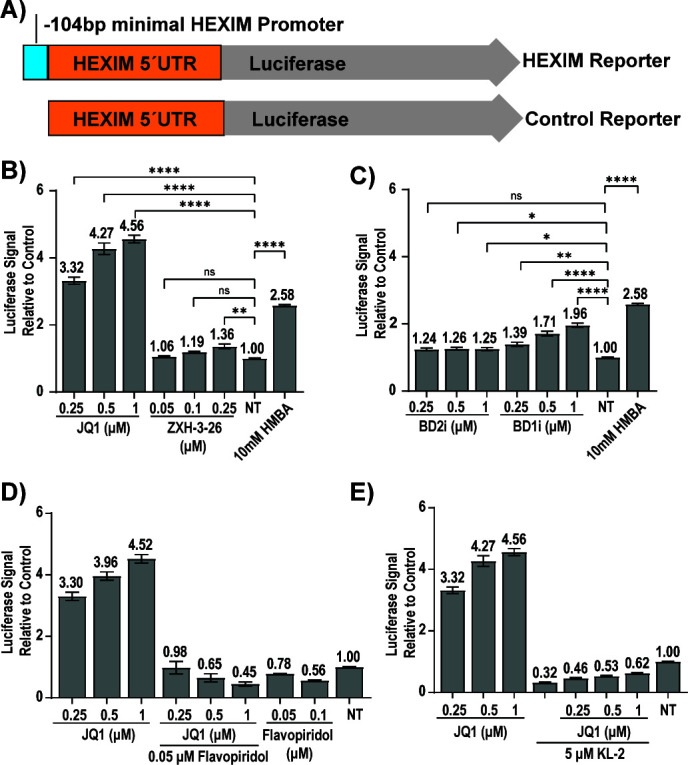
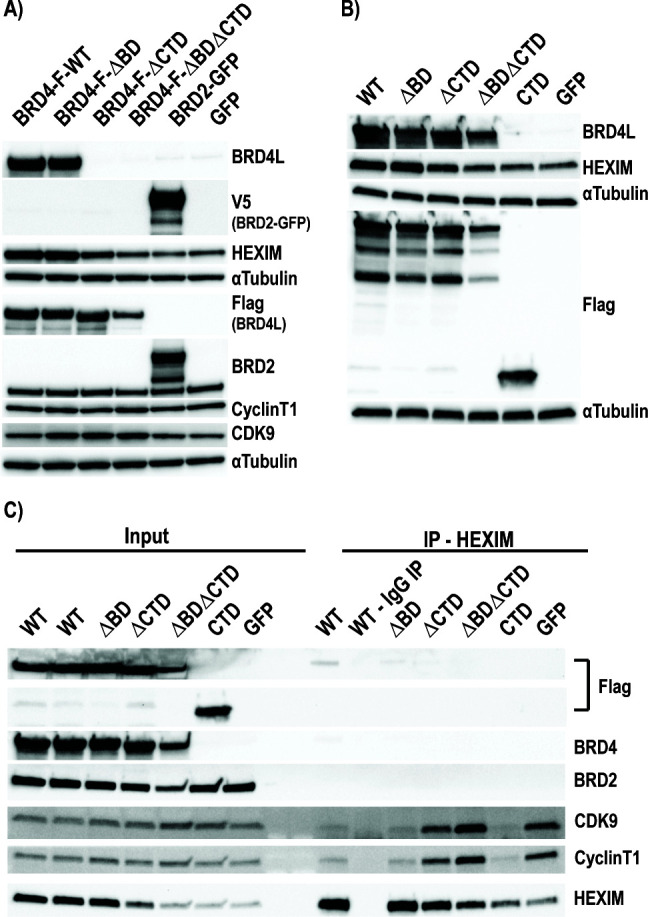
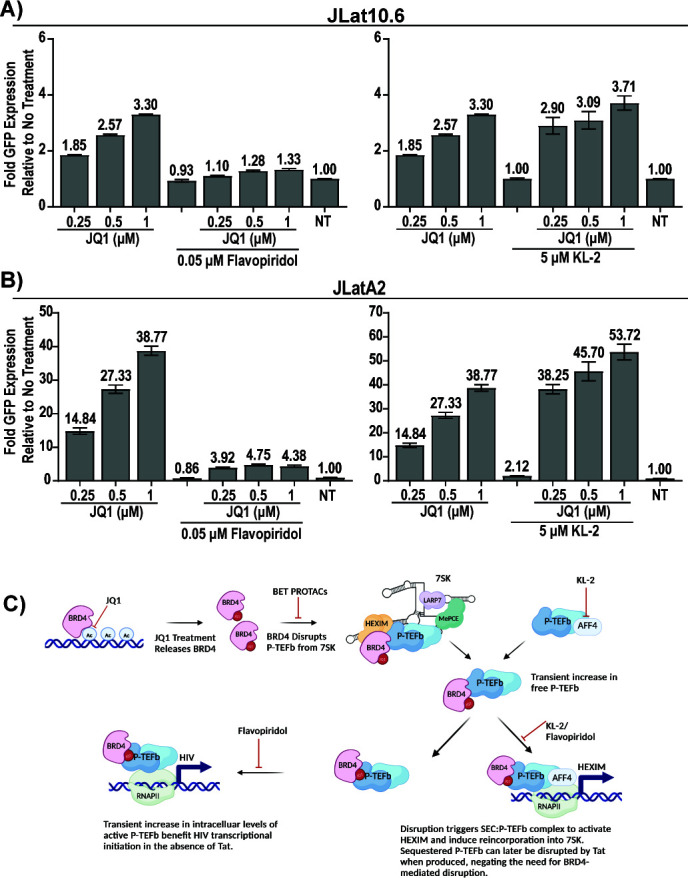
HIV cure strategies that aim to induce viral reactivation for immune clearance leverage latency reversal agents to modulate host pathways which directly or indirectly facilitate viral reactivation. Inhibition of bromo and extra-terminal domain (BET) family member BRD4 reverses HIV latency, but enthusiasm for the use of BET inhibitors in HIV cure studies is tempered by concerns over inhibition of other BET family members and dose-limiting toxicities in oncology trials. Here, we evaluated the potential for bivalent chemical degraders targeted to the BET family as alternative latency reversal agents. We observed that despite highly potent and selective BRD4 degradation in primary CD4+ T-cells from ART-suppressed donors, BRD4 degraders failed to induce latency reversal as compared to BET inhibitors. Furthermore, BRD4 degraders failed to mimic previously observed synergistic HIV reactivation between BET inhibitors and an activator of the non-canonical NF-κB pathway. Mechanistic investigation of this discrepancy revealed that latency reversal by BET inhibitors is not related to the abatement of competition between Tat and BRD4 for P-TEFb, but rather the ability of BRD4 to disrupt 7SK and increase the levels of free P-TEFb. This activity is dependent on the shift of BRD4 from chromatin-bound to soluble and retargeting of P-TEFb to chromatin, which is dependent on intact BRD4 but independent of the bromodomains.
Importance: Multiple factors and pathways contribute to the maintenance of HIV latency, including bromo and extra-terminal domain (BET) family member BRD4. While small molecule inhibitors of the BET family result in latency reversal, enthusiasm for the use of BET inhibitors in HIV cure is limited due to toxicity concerns. We examined BRD4-selective chemical degraders as alternatives to BET inhibitors but found two robust degraders failed to induce latency reversal. We observed key differences in the ability of BET inhibitors versus BET degraders to disrupt P-TEFb, a key cellular activator of transcription and a complex required for HIV reactivation. We present a new model for the role of BRD4 in HIV latency and propose that BRD4 be reconsidered as an activator rather than a repressor of HIV transcription in the context of HIV cure strategies.
Keywords: BRD4; HIV; P-TEFb; chemical degraders; latency reversal.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Integrator complex subunit 12 knockout overcomes a transcriptional block to HIV latency reversal.Elife. 2025 Apr 10;13:RP103064. doi: 10.7554/eLife.103064. Elife. 2025. PMID: 40207620 Free PMC article.
-
Inhibition of ALKBH5 demethylase of m6A pathway potentiates HIV-1 reactivation from latency.Virol J. 2025 Apr 28;22(1):124. doi: 10.1186/s12985-025-02744-4. Virol J. 2025. PMID: 40296171 Free PMC article.
-
Tannic acid reactivates HIV-1 latency by mediating CBX4 degradation.J Virol. 2025 Jan 31;99(1):e0117324. doi: 10.1128/jvi.01173-24. Epub 2024 Dec 18. J Virol. 2025. PMID: 39692477 Free PMC article.
-
Structured treatment interruptions (STI) in chronic unsuppressed HIV infection in adults.Cochrane Database Syst Rev. 2006 Jul 19;2006(3):CD006148. doi: 10.1002/14651858.CD006148. Cochrane Database Syst Rev. 2006. PMID: 16856117 Free PMC article.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2021 Apr 19;4(4):CD011535. doi: 10.1002/14651858.CD011535.pub4. Cochrane Database Syst Rev. 2021. Update in: Cochrane Database Syst Rev. 2022 May 23;5:CD011535. doi: 10.1002/14651858.CD011535.pub5. PMID: 33871055 Free PMC article. Updated.
Cited by
-
Integrator complex subunit 12 knockout overcomes a transcriptional block to HIV latency reversal.bioRxiv [Preprint]. 2025 Feb 19:2024.08.30.610517. doi: 10.1101/2024.08.30.610517. bioRxiv. 2025. Update in: Elife. 2025 Apr 10;13:RP103064. doi: 10.7554/eLife.103064. PMID: 39257755 Free PMC article. Updated. Preprint.
-
Integrator complex subunit 12 knockout overcomes a transcriptional block to HIV latency reversal.Elife. 2025 Apr 10;13:RP103064. doi: 10.7554/eLife.103064. Elife. 2025. PMID: 40207620 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
