

A novel strategy for sorafenib-resistant hepatocellular carcinoma: autotaxin Inhibition by PF-8380
- PMID: 40082280
- PMCID: PMC11906571
- DOI: 10.1007/s00432-025-06156-3
A novel strategy for sorafenib-resistant hepatocellular carcinoma: autotaxin Inhibition by PF-8380
Abstract
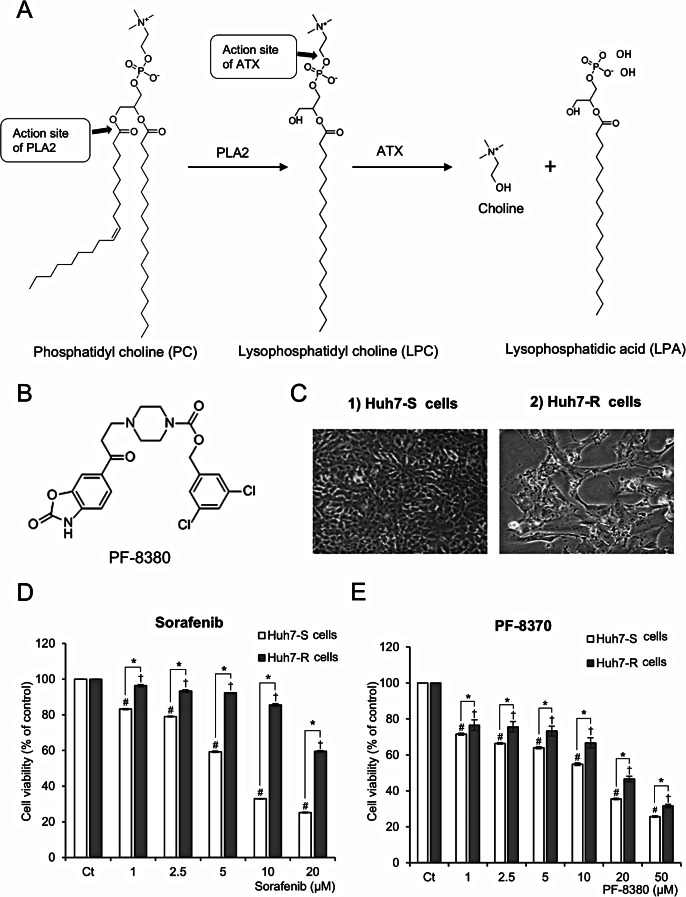
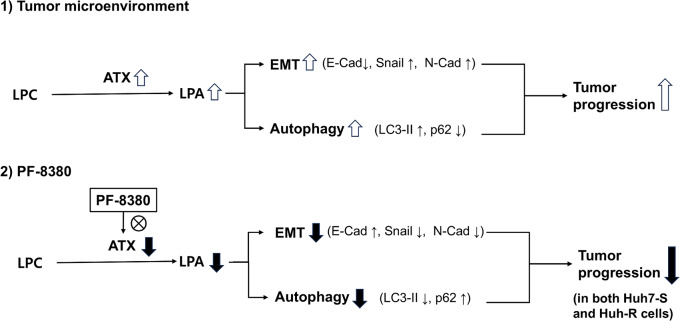
By inhibiting the conversion of lysophosphatidylcholine into lysophosphatidic acid, a process pivotal to tumor progression, the autotaxin (ATX) inhibitor PF-8380 offers a new anticancer therapeutic strategy, distinct from the action mechanism of sorafenib. This study explored the potential anticancer effects of the PF-8380 on hepatocellular carcinoma (HCC) cells, especially sorafenib-resistant strains. The investigation included both in vitro and in vivo experiments to evaluate the impact of PF-8380 treatment on epithelial-mesenchymal transition (EMT) and autophagy markers. An orthotopic HCC model served as the in vivo platform. PF-8380 showed a significant reduction in cell viability in both sorafenib-susceptible and resistant HCC cells. It effectively altered EMT by increasing E-cadherin and reducing Snail levels, and inhibited autophagy, as indicated by changes in LC3 and p62 markers. These effects were consistently observed in the orthotopic HCC mouse model, reinforcing PF-8380's potential as a dual inhibitor of EMT and autophagy in HCC treatment. Our research indicates that PF-8380 could provide substantial therapeutic benefits in the treatment of HCC, even in cases resistant to sorafenib, primarily by suppressing both EMT and autophagy processes.
Keywords: Autophagy; Autotaxin inhibitor; Epithelial-mesenchymal transition (EMT); Hepatocellular carcinoma; Sorafenib.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethical approval: The animal studies adhered to the guidelines of the Institute for Laboratory Animal Research, Korea (IRB No: CUMC-2020-0211-06). Competing interests: The authors declare no competing interests.
Figures




References
-
- Chen L, Zhang J, Deng X, Liu Y, Yang X, Wu Q, Yu C (2017) Lysophosphatidic acid directly induces macrophage-derived foam cell formation by blocking the expression of SRBI. Biochem Biophys Res Commun 491:587–594. 10.1016/j.bbrc.2017.07.159 - PubMed
-
- Fukushima K, Takahashi K, Yamasaki E, Onishi Y, Fukushima N, Honoki K, Tsujiuchi T (2017) Lysophosphatidic acid signaling via LPA(1) and LPA(3) regulates cellular functions during tumor progression in pancreatic cancer cells. Exp Cell Res 352:139–145. 10.1016/j.yexcr.2017.02.007 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
