

A systematic benchmark of Nanopore long-read RNA sequencing for transcript-level analysis in human cell lines
- PMID: 40082608
- PMCID: PMC11978509
- DOI: 10.1038/s41592-025-02623-4
A systematic benchmark of Nanopore long-read RNA sequencing for transcript-level analysis in human cell lines
Abstract
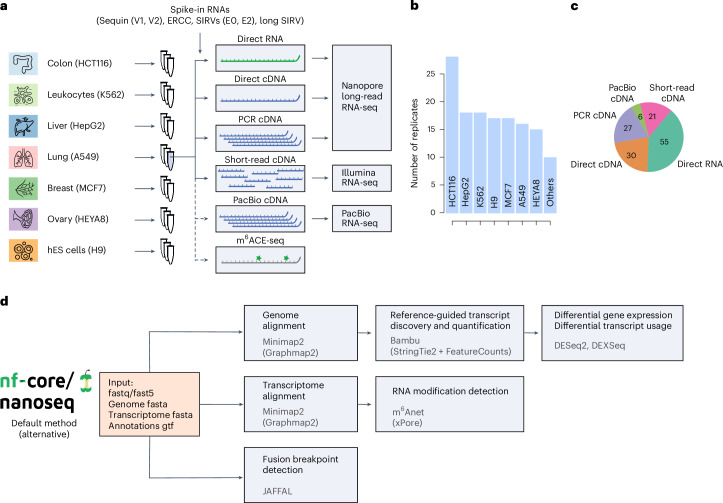
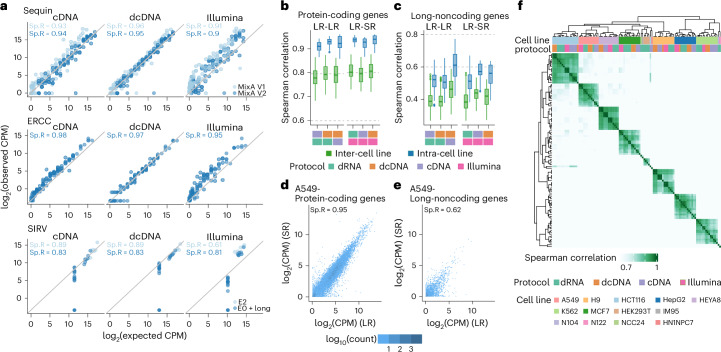
The human genome contains instructions to transcribe more than 200,000 RNAs. However, many RNA transcripts are generated from the same gene, resulting in alternative isoforms that are highly similar and that remain difficult to quantify. To evaluate the ability to study RNA transcript expression, we profiled seven human cell lines with five different RNA-sequencing protocols, including short-read cDNA, Nanopore long-read direct RNA, amplification-free direct cDNA and PCR-amplified cDNA sequencing, and PacBio IsoSeq, with multiple spike-in controls, and additional transcriptome-wide N6-methyladenosine profiling data. We describe differences in read length, coverage, throughput and transcript expression, reporting that long-read RNA sequencing more robustly identifies major isoforms. We illustrate the value of the SG-NEx data to identify alternative isoforms, novel transcripts, fusion transcripts and N6-methyladenosine RNA modifications. Together, the SG-NEx data provide a comprehensive resource enabling the development and benchmarking of computational methods for profiling complex transcriptional events at isoform-level resolution.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: J.G. received travel and accommodation expenses to speak at the Oxford Nanopore Community Meeting 2018. N.M.D. has previously received travel and accommodation expenses from Oxford Nanopore Technologies. H.G. has previously received travel and accommodation expenses from Oxford Nanopore Technologies. M.S. has been jointly funded by Oxford Nanopore Technologies and AI Singapore for the project AI-driven De Novo Diploid Assembler and has received travel funds to speak at events hosted by Oxford Nanopore Technologies. W.S.S.G. owns shares in Oxford Nanopore Technologies. The other authors declare no competing interests.
Figures




References
-
- Demircioğlu, D. et al. A pan-cancer transcriptome analysis reveals pervasive regulation through alternative promoters. Cell178, 1465–1477 (2019). - PubMed
MeSH terms
Grants and funding
- OFIRG20nov-0108/MOH | National Medical Research Council (NMRC)
- OFIRG16nov019/MOH | National Medical Research Council (NMRC)
- GNT2016547/Department of Health | National Health and Medical Research Council (NHMRC)
- NHMRC GNT1196256/Department of Health | National Health and Medical Research Council (NHMRC)
- MOE-MOET32023-0002/Ministry of Education - Singapore (MOE)
LinkOut - more resources
Full Text Sources
