

Optimizing Xenium In Situ data utility by quality assessment and best-practice analysis workflows
- PMID: 40082609
- PMCID: PMC11978515
- DOI: 10.1038/s41592-025-02617-2
Optimizing Xenium In Situ data utility by quality assessment and best-practice analysis workflows
Abstract
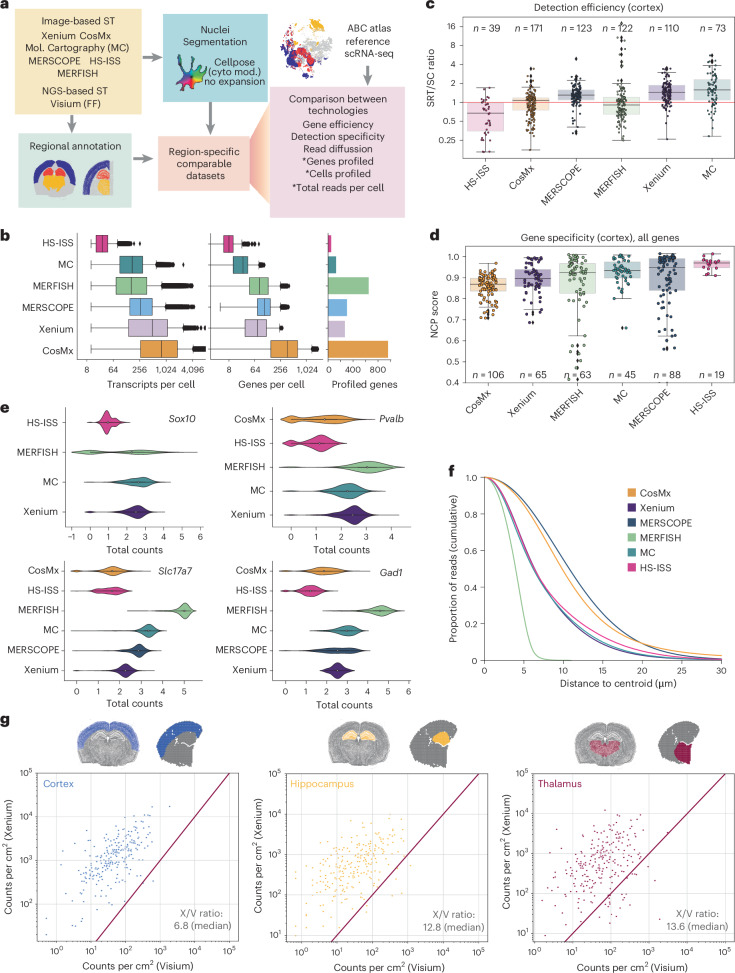
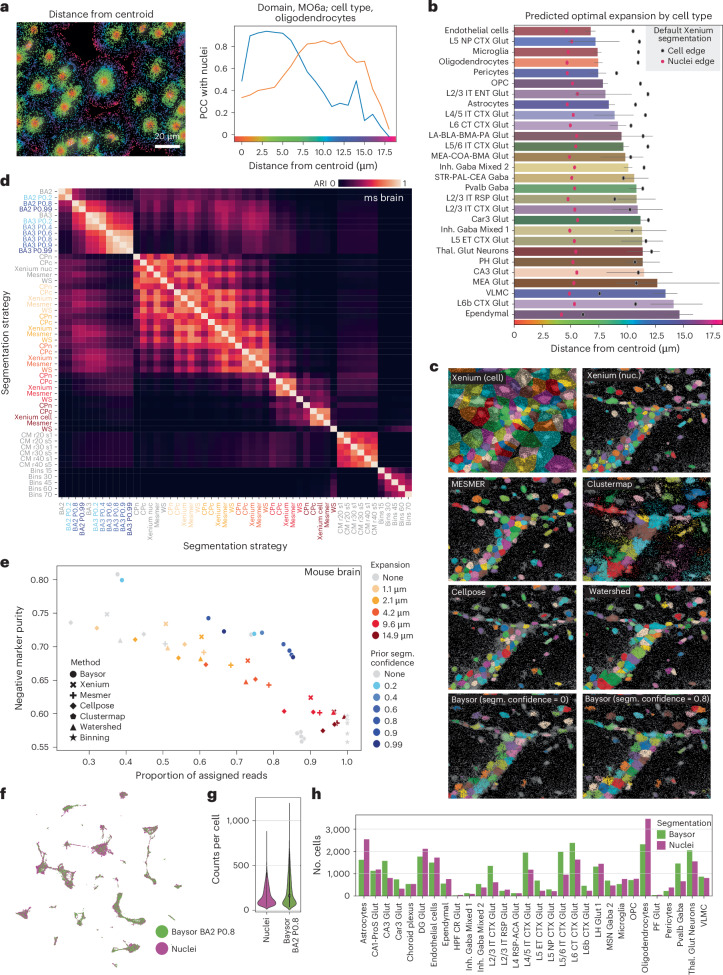
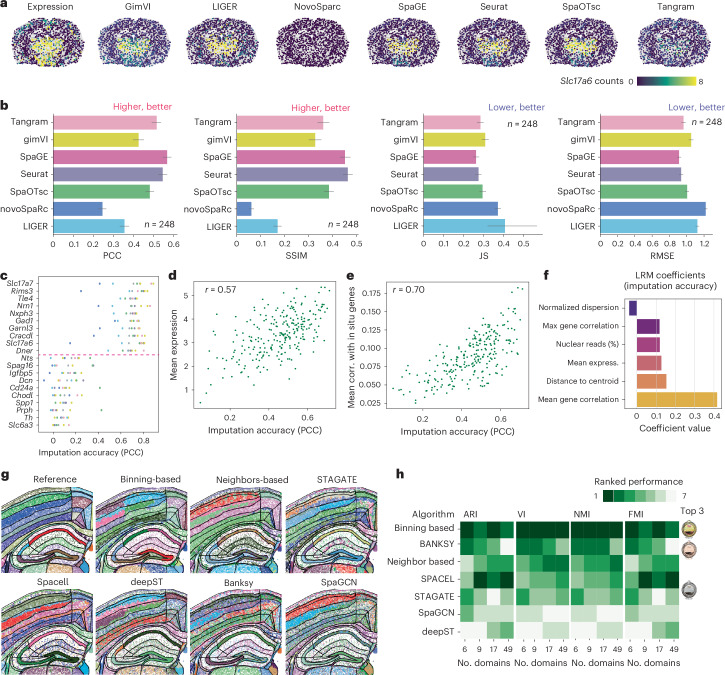
The Xenium In Situ platform is a new spatial transcriptomics product commercialized by 10x Genomics, capable of mapping hundreds of genes in situ at subcellular resolution. Given the multitude of commercially available spatial transcriptomics technologies, recommendations in choice of platform and analysis guidelines are increasingly important. Herein, we explore 25 Xenium datasets generated from multiple tissues and species, comparing scalability, resolution, data quality, capacities and limitations with eight other spatially resolved transcriptomics technologies and commercial platforms. In addition, we benchmark the performance of multiple open-source computational tools, when applied to Xenium datasets, in tasks including preprocessing, cell segmentation, selection of spatially variable features and domain identification. This study serves as an independent analysis of the performance of Xenium, and provides best practices and recommendations for analysis of such datasets.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: M.N. was an advisor for 10x Genomics when this manuscript was initially submitted, but he is no longer involved in any advisory role for the company. F.J.T. consults for Immunai, Singularity Bio, CytoReason and Omniscope, and has ownership interest in Dermagnostix and Cellarity. M.D.L. contracted for the Chan Zuckerberg Initiative and received speaker fees from Pfizer and Janssen Pharmaceuticals. S.M.S., C.M.L. and M.G. are co-founders of Spatialist, a data-analysis company focused on spatial omics. The remaining authors declare no competing interests.
Figures









References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
