

Feature selection methods affect the performance of scRNA-seq data integration and querying
- PMID: 40082610
- PMCID: PMC11978513
- DOI: 10.1038/s41592-025-02624-3
Feature selection methods affect the performance of scRNA-seq data integration and querying
Abstract
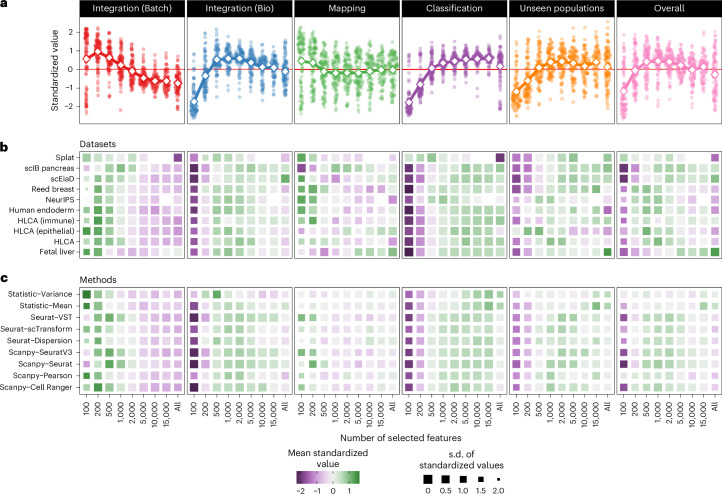
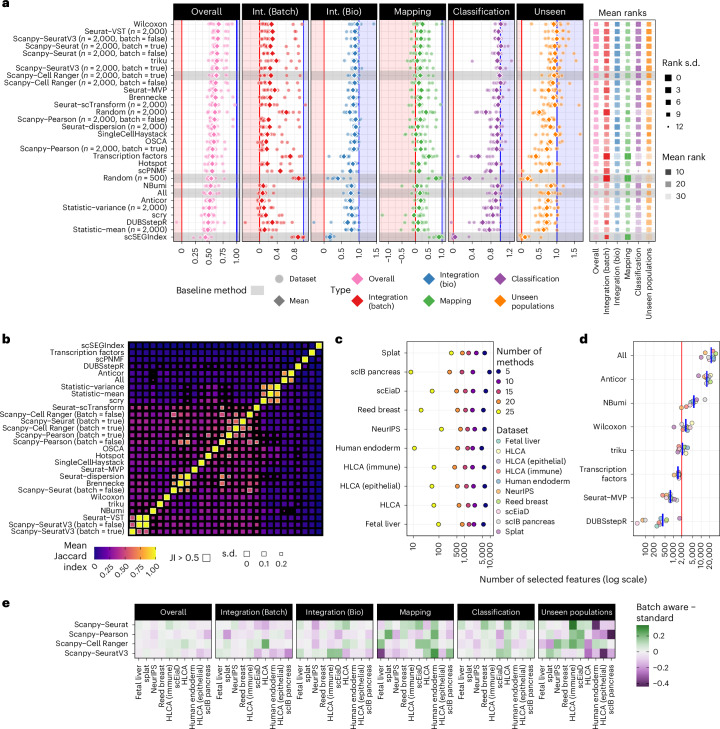
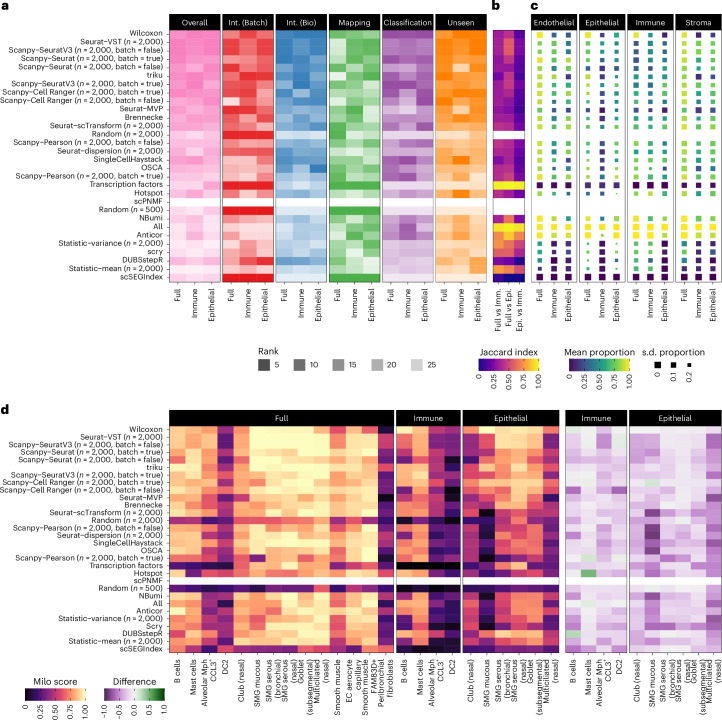
The availability of single-cell transcriptomics has allowed the construction of reference cell atlases, but their usefulness depends on the quality of dataset integration and the ability to map new samples. Previous benchmarks have compared integration methods and suggest that feature selection improves performance but have not explored how best to select features. Here, we benchmark feature selection methods for single-cell RNA sequencing integration using metrics beyond batch correction and preservation of biological variation to assess query mapping, label transfer and the detection of unseen populations. We reinforce common practice by showing that highly variable feature selection is effective for producing high-quality integrations and provide further guidance on the effect of the number of features selected, batch-aware feature selection, lineage-specific feature selection and integration and the interaction between feature selection and integration models. These results are informative for analysts working on large-scale tissue atlases, using atlases or integrating their own data to tackle specific biological questions.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: F.J.T. consults for Immunai, Singularity Bio, CytoReason and Cellarity and has an ownership interest in Dermagnostix and Cellarity. A.F. is currently an employee of CytoReason. L.Z. has consulted for Lamin Labs, was an employee of iOmx Therapeutics and is currently an employee of Data Intuitive. R.K.-R. has consulted for iuvando Health. M.D.L. consults for CatalYm, has contracted for the Chan Zuckerberg Initiative and has received speaker fees from Pfizer and Janssen Pharmaceuticals. The other authors declare no competing interests.
Figures











References
-
- Regev, A. et al. Human cell atlas meeting participants. The human cell atlas. eLife10.7554/elife.27041 (2017).
-
- Mereu, E. et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nat. Biotechnol.38, 747–755 (2020). - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
