

Rescue of the disease-associated phenotype in CRISPR-corrected hiPSCs as a therapeutic approach for inherited retinal dystrophies
- PMID: 40083649
- PMCID: PMC11903799
- DOI: 10.1016/j.omtn.2025.102482
Rescue of the disease-associated phenotype in CRISPR-corrected hiPSCs as a therapeutic approach for inherited retinal dystrophies
Abstract
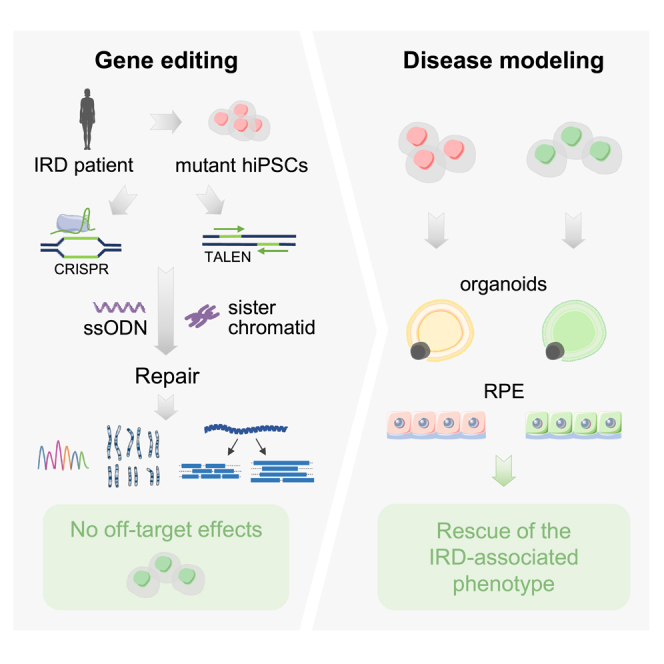
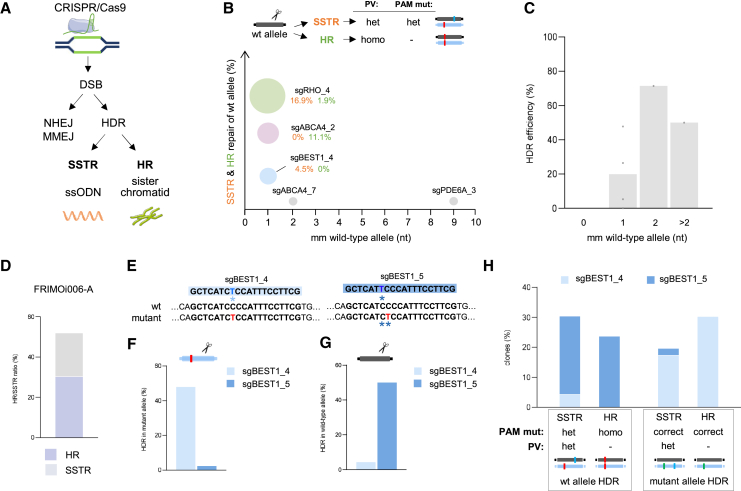
Inherited retinal dystrophies (IRDs), such as retinitis pigmentosa and Stargardt disease, are a group of rare diseases caused by mutations in more than 300 genes that currently have no treatment in most cases. They commonly trigger blindness and other ocular affectations due to retinal cell degeneration. Gene editing has emerged as a promising and powerful strategy for the development of IRD therapies, allowing the permanent correction of pathogenic variants. Using clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 and transcription activator-like effector nucleases (TALEN) gene-editing tools, we precisely corrected seven hiPS cell lines derived from IRD patients carrying mutations in ABCA4, BEST1, PDE6A, PDE6C, RHO, or USH2A. Homozygous mutations and point insertions/deletions resulted in the highest homology-directed repair efficiencies, with at least half of the clones repaired properly without off-target effects. Strikingly, correction of a heterozygous pathogenic variant was achieved using the wild-type allele of the patient as the template for DNA repair. These results suggest the unexpected potential application of CRISPR as a donor template-free strategy for single-nucleotide modifications. Additionally, the corrected clones exhibited a reversion of the disease-associated phenotype in retinal cellular models. These data strengthen the study and application of gene editing-based approaches for IRD treatment.
Keywords: Best disease; CRISPR; MT: RNA/DNA Editing; TALEN; cell therapy; gene editing; gene therapy; inherited retinal dystrophies; retinitis pigmentosa; sister chromatid.
© 2025 The Author(s).
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
-
- Schneider N., Sundaresan Y., Gopalakrishnan P., Beryozkin A., Hanany M., Levanon E.Y., Banin E., Ben-Aroya S., Sharon D. Inherited retinal diseases: Linking genes, disease-causing variants, and relevant therapeutic modalities. Prog. Retin. Eye Res. 2022;89 doi: 10.1016/j.preteyeres.2021.101029. - DOI - PubMed
-
- Biswas P., Villanueva A.L., Soto-Hermida A., Duncan J.L., Matsui H., Borooah S., Kurmanov B., Richard G., Khan S.Y., Branham K., et al. Deciphering the genetic architecture and ethnographic distribution of IRD in three ethnic populations by whole genome sequence analysis. PLoS Genet. 2021;17 doi: 10.1371/journal.pgen.1009848. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
