

Zebrafish and cellular models of SELENON-Congenital myopathy exhibit novel embryonic and metabolic phenotypes
- PMID: 40087793
- PMCID: PMC11909958
- DOI: 10.1186/s13395-025-00376-4
Zebrafish and cellular models of SELENON-Congenital myopathy exhibit novel embryonic and metabolic phenotypes
Abstract
Background: SELENON-Congenital Myopathy (SELENON-CM) is a rare congenital myopathy caused by mutations of the SELENON gene characterized by axial muscle weakness and progressive respiratory insufficiency. Muscle histopathology may be non-specific, but commonly includes multiminicores or a dystrophic pattern. The SELENON gene encodes selenoprotein N (SelN), a selenocysteine-containing redox enzyme located in the endo/sarcoplasmic reticulum membrane where it colocalizes with mitochondria-associated membranes. However, the molecular mechanism(s) by which SelN deficiency cause SELENON-CM remain poorly understood. A hurdle is the lack of cellular and animal models that show easily assayable phenotypes.
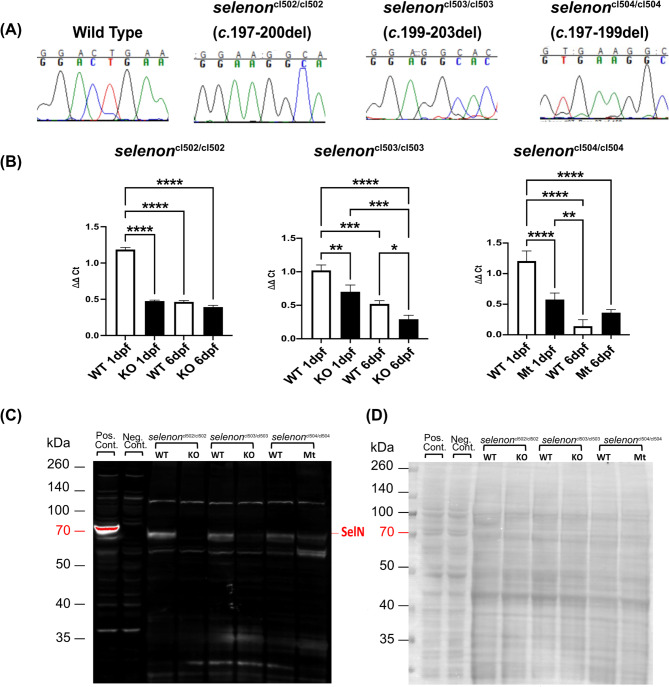
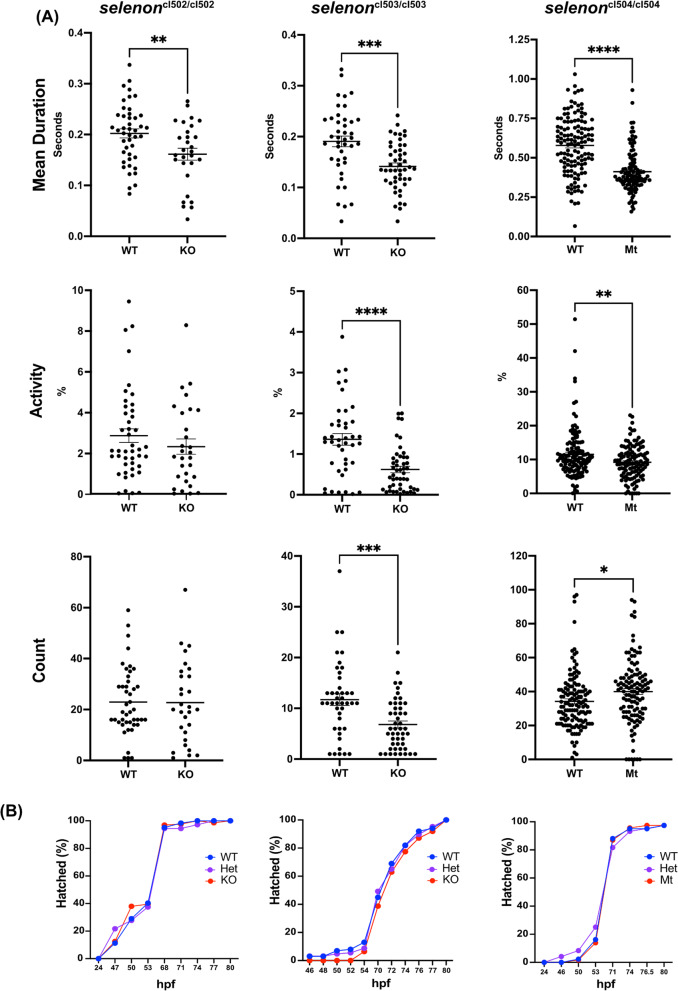
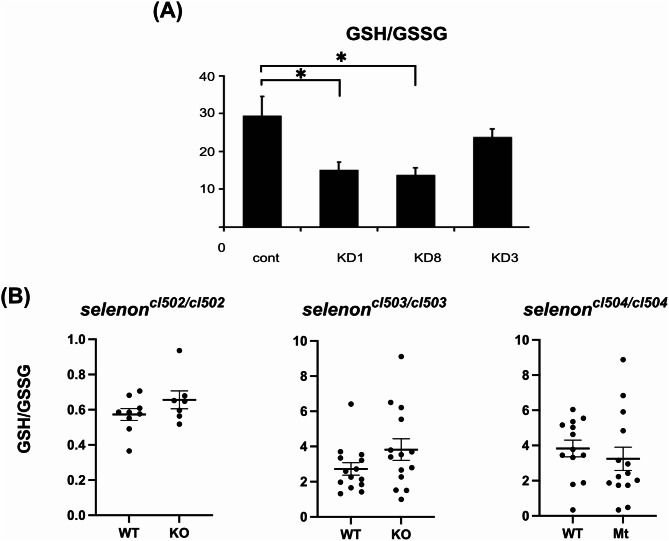
Methods: Using CRISPR-Cas9 we generated three zebrafish models of SELENON-CM, which were then studied by spontaneous coiling, hatching, and activity assays. We also performed selenon coexpression analysis using a single cell RNAseq zebrafish embryo-atlas. SelN-deficient myoblasts were generated and assayed for glutathione, reactive oxygen species, carbonylation, and nytrosylation levels. Finally, we tested Selenon-deficient myoblasts' metabolism using a Seahorse cell respirometer.
Results: We report deep-phenotyping of SelN-deficient zebrafish and muscle cells. SelN-deficient zebrafish exhibit changes in embryonic muscle function and swimming activity in larvae. Analysis of single cell RNAseq data in a zebrafish embryo-atlas revealed coexpression of selenon and genes involved in the glutathione redox pathway. SelN-deficient zebrafish and mouse myoblasts exhibit altered glutathione and redox homeostasis, as well as abnormal patterns of energy metabolism, suggesting roles for SelN in these functions.
Conclusions: These data demonstrate a role for SelN in zebrafish early development and myoblast metabolism and provide a basis for cellular and animal model assays for SELENON-CM.
Keywords: Congenital myopathy; Multiminicore myopathy; Rigid spine muscular dystrophy; Selenoprotein N; Zebrafish model.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: AHB receives consulting income from Kate Therapeutics, Astellas Pharma, Roche Pharmaceuticals, GLG Inc, and Guidepoint Global, and has equity in Kate Therapeutics and Kinea Bio. For all other authors no competing interests are declared.
Figures



Update of
-
Zebrafish and cellular models of SELENON-Related Myopathy exhibit novel embryonic and metabolic phenotypes.bioRxiv [Preprint]. 2024 Feb 26:2024.02.26.581979. doi: 10.1101/2024.02.26.581979. bioRxiv. 2024. Update in: Skelet Muscle. 2025 Mar 15;15(1):7. doi: 10.1186/s13395-025-00376-4. PMID: 38464009 Free PMC article. Updated. Preprint.
References
-
- Moghadaszadeh B, Petit N, Jaillard C, Brockington M, Roy SQ, Merlini L, et al. Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nat Genet. 2001;29:17–8. - PubMed
-
- Ferreiro A, Quijano-Roy S, Pichereau C, Moghadaszadeh B, Goemans N, Bönnemann G, et al. Mutations of the Selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet Cell Press. 2002;71:739–49. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
