

Benchmarking of Quantitative Proteomics Workflows for Limited Proteolysis Mass Spectrometry
- PMID: 40089063
- PMCID: PMC12022698
- DOI: 10.1016/j.mcpro.2025.100945
Benchmarking of Quantitative Proteomics Workflows for Limited Proteolysis Mass Spectrometry
Abstract
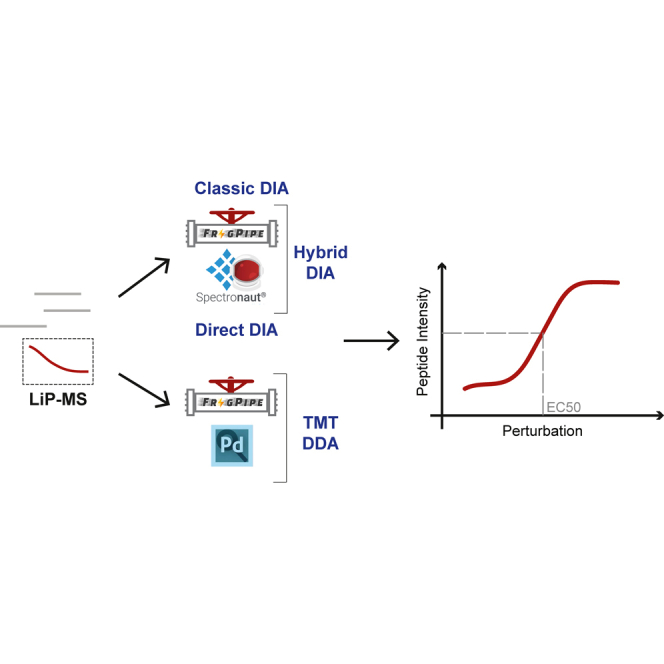
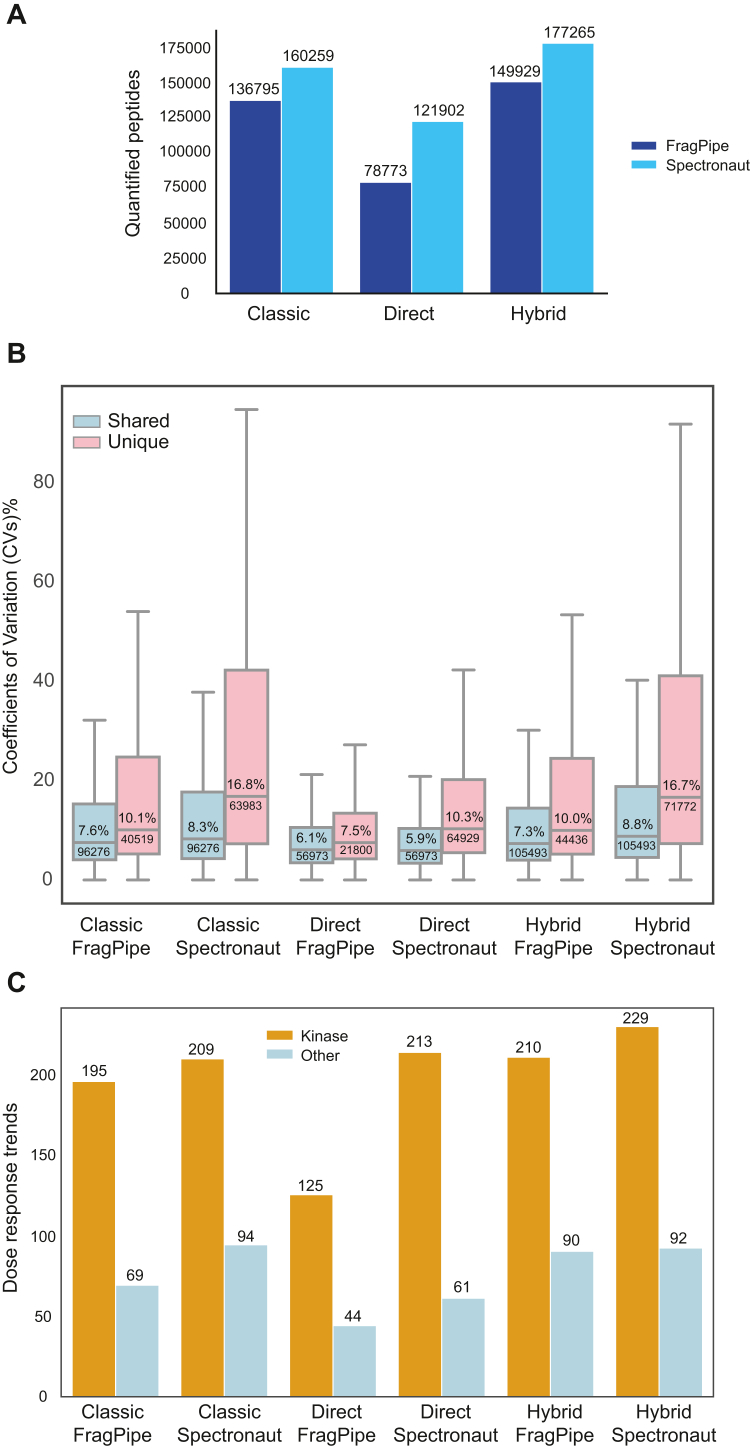
Limited proteolysis coupled with mass spectrometry (LiP-MS) has emerged as a powerful technique for detecting protein structural changes and drug-protein interactions on a proteome-wide scale. However, there is no consensus on the best quantitative proteomics workflow for analyzing LiP-MS data. In this study, we comprehensively benchmarked two major quantification approaches-data-independent acquisition (DIA) and tandem mass tag (TMT) isobaric labeling-in combination with LiP-MS, using a drug-target deconvolution assay as a model system. Our results show that while TMT labeling enabled the quantification of more peptides and proteins with lower coefficients of variation, DIA-MS exhibited greater accuracy in identifying true drug targets and stronger dose-response correlation in peptides of protein targets. Additionally, we evaluated the performance of freely available (FragPipe) versus commercial (Spectronaut) software tools for DIA-MS analysis, revealing that the choice between precision (FragPipe) and sensitivity (Spectronaut) largely depends on the specific experimental context. Our findings underscore the importance of selecting the appropriate LiP-MS quantification strategy based on the study objectives. This work provides valuable guidelines for researchers in structural proteomics and drug discovery, and highlights how advancements in mass spectrometry instrumentation, such as the Astral mass spectrometer, may further improve sensitivity and protein sequence coverage, potentially reducing the need for TMT labeling.
Keywords: DIA software benchmarking; DIA-MS; FAIMS; FragPipe; LiP-MS; TMT; structural proteomics.
Copyright © 2025 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare no competing interests.
Figures



References
-
- Feng Y., De Franceschi G., Kahraman A., Soste M., Melnik A., Boersema P.J., et al. Global analysis of protein structural changes in complex proteomes. Nat. Biotechnol. 2014;32:1036–1044. - PubMed
-
- Adhikari J., Fitzgerald M.C. SILAC-pulse proteolysis: a mass spectrometry-based method for discovery and cross-validation in proteome-wide studies of ligand binding. J. Am. Soc. Mass Spectrom. 2014;25:2073–2083. - PubMed
-
- Park C., Marqusee S. Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding. Nat. Methods. 2005;2:207–212. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
