

Molecular glues that inhibit deubiquitylase activity and inflammatory signaling
- PMID: 40097626
- PMCID: PMC7617869
- DOI: 10.1038/s41594-025-01517-5
Molecular glues that inhibit deubiquitylase activity and inflammatory signaling
Abstract
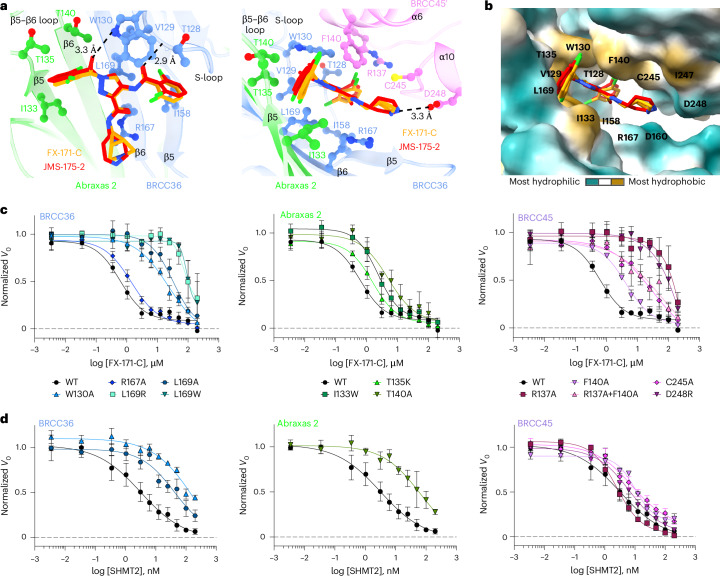
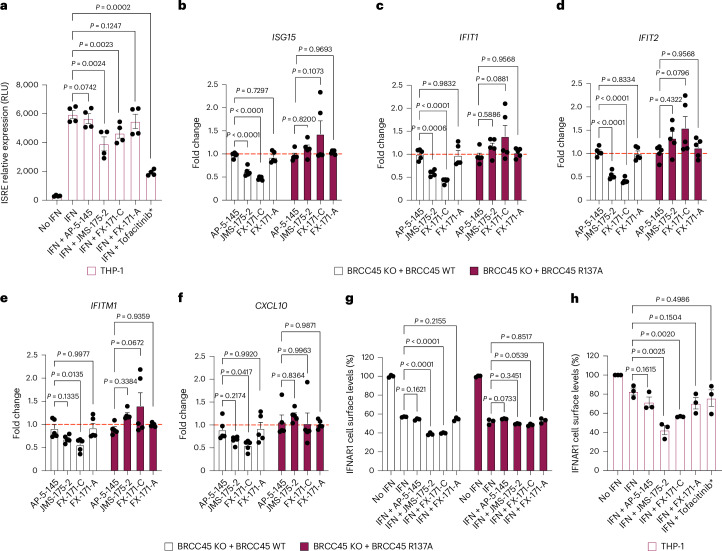
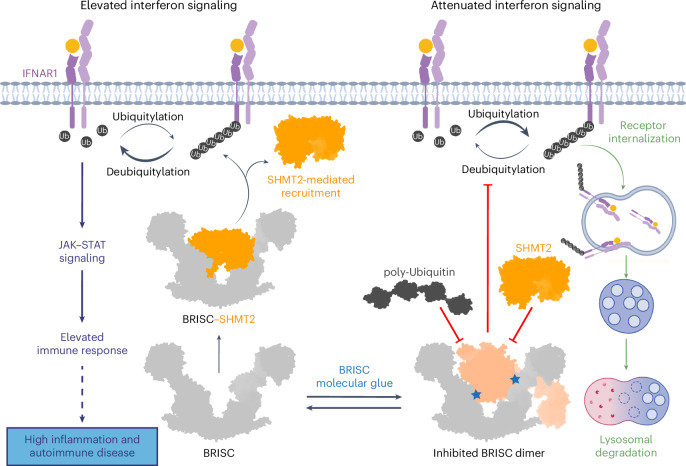
Deubiquitylases (DUBs) are crucial in cell signaling and are often regulated by interactions within protein complexes. The BRCC36 isopeptidase complex (BRISC) regulates inflammatory signaling by cleaving K63-linked polyubiquitin chains on type I interferon receptors (IFNAR1). As a Zn2+-dependent JAMM/MPN (JAB1, MOV34, MPR1, Pad1 N-terminal) DUB, BRCC36 is challenging to target with selective inhibitors. Here, we discover first-in-class inhibitors, termed BRISC molecular glues (BLUEs), which stabilize a 16-subunit human BRISC dimer in an autoinhibited conformation, blocking active sites and interactions with the targeting subunit, serine hydroxymethyltransferase 2. This unique mode of action results in selective inhibition of BRISC over related complexes with the same catalytic subunit, splice variants and other JAMM/MPN DUBs. BLUE treatment reduced interferon-stimulated gene expression in cells containing wild-type BRISC and this effect was abolished when using structure-guided, inhibitor-resistant BRISC mutants. Additionally, BLUEs increase IFNAR1 ubiquitylation and decrease IFNAR1 surface levels, offering a potential strategy to mitigate type I interferon-mediated diseases. Our approach also provides a template for designing selective inhibitors of large protein complexes by promoting rather than blocking protein-protein interactions.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: E.Z., R.G., J.M.S. and F.S. are coinventors on a patent to use BRISC inhibitors as therapeutics (WO2024115713A1). J.M.S. owns equity in Alliance Discovery and the Barer Institute and consults for Syndeavor Therapeutics. The remaining authors declare no competing interests.
Figures













Update of
-
Molecular glues that inhibit deubiquitylase activity and inflammatory signalling.bioRxiv [Preprint]. 2024 Nov 15:2024.09.07.611787. doi: 10.1101/2024.09.07.611787. bioRxiv. 2024. Update in: Nat Struct Mol Biol. 2025 Sep;32(9):1812-1824. doi: 10.1038/s41594-025-01517-5. PMID: 39282282 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
