

Developing a more accurate population frequency of Marfan syndrome from predicted pathogenic FBN1 variants in the gnomAD cohorts
- PMID: 40102521
- PMCID: PMC11920421
- DOI: 10.1038/s41598-025-93832-6
Developing a more accurate population frequency of Marfan syndrome from predicted pathogenic FBN1 variants in the gnomAD cohorts
Abstract
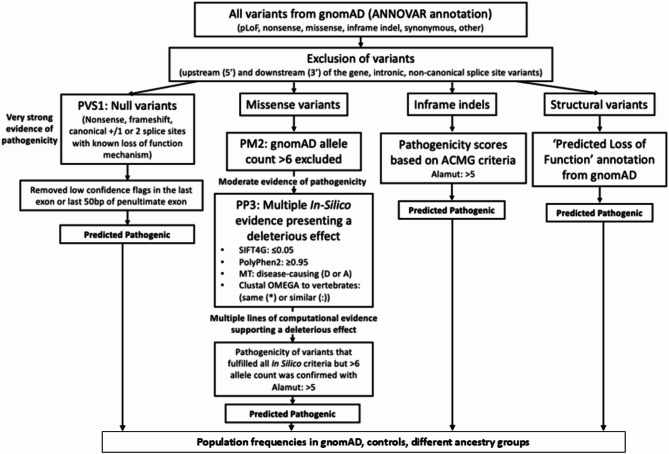
Marfan syndrome is an autosomal dominantly (AD)-inherited disease that results from pathogenic variants in the Fibrillin 1 (FBN1) gene, and is characterised by tall stature, elongated limbs and digits, lens abnormalities and aortic root dilatation, aneurysms and dissection, but milder forms also occur. Radiological imaging suggests that Marfan syndrome affects between one in 3000 and 5000 of the population. The aim of this study was to determine the population frequency of Marfan syndrome from the number of predicted pathogenic FBN1 changes found in a normal database. FBN1 variants were downloaded from gnomAD v2.1.1 and annotated with ANNOVAR. The population frequency was determined from the number of pathogenic null and structural variants, and the number of predicted pathogenic missense changes classified by rarity and computational scores. This population frequency was then compared with the frequencies in the control subset, and from gnomAD variants assessed as Pathogenic or Likely pathogenic in the ClinVar or LOVD databases. Our strategy identified predicted pathogenic FBN1 variants in one in 416 individuals, which was confirmed in the control subset (one in 356, p NS). Predicted pathogenic variants were most common in East Asian people (one in 243, p < 0.0001) and least common in Ashkenazim (one in 5185, p = 0.0082). The population frequencies based on pathogenic variants in the ClinVar or LOVD databases were one in 718 and one in 1014 respectively. Null variants which are associated with aortic aneurysms affected only one in 8624. Thus, Marfan syndrome is more common than previously recognised. Emergency departments and cardiac clinics in particular should be aware of undiagnosed Marfan syndrome and its cardiac risks, but many of those affected still have a milder phenotype.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Autosomal dominant Marfan syndrome caused by a previously reported recessive FBN1 variant.Mol Genet Genomic Med. 2019 Feb;7(2):e00518. doi: 10.1002/mgg3.518. Epub 2018 Nov 28. Mol Genet Genomic Med. 2019. PMID: 30485715 Free PMC article.
-
Genotype and clinical phenotype of children with Marfan syndrome in Southeastern Anatolia.Eur J Pediatr. 2024 Aug;183(8):3219-3232. doi: 10.1007/s00431-024-05579-3. Epub 2024 May 3. Eur J Pediatr. 2024. PMID: 38700693 Free PMC article.
-
Re-evaluation of a Fibrillin-1 Gene Variant of Uncertain Significance Using the ClinGen Guidelines.Ann Lab Med. 2024 May 1;44(3):271-278. doi: 10.3343/alm.2023.0152. Epub 2023 Oct 16. Ann Lab Med. 2024. PMID: 37840311 Free PMC article.
-
A novel fibrillin-1 gene missense mutation associated with neonatal Marfan syndrome: a case report and review of the mutation spectrum.BMC Pediatr. 2016 Apr 30;16:60. doi: 10.1186/s12887-016-0598-6. BMC Pediatr. 2016. PMID: 27138491 Free PMC article. Review.
-
FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders.Gene. 2016 Oct 10;591(1):279-291. doi: 10.1016/j.gene.2016.07.033. Epub 2016 Jul 18. Gene. 2016. PMID: 27437668 Free PMC article. Review.
References
-
- Dietz, H. FBN1-Related Marfan Syndrome. In GeneReviews((R)) (eds Adam, M. P. et al.) (1993). - PubMed
-
- Mannucci, L. et al. Mutation analysis of the FBN1 gene in a cohort of patients with Marfan Syndrome: A 10-year single center experience. Clin. Chim. Acta501, 154–164 (2020). - PubMed
-
- Boucek, R. J., Noble, N. L., Gunja-Smith, Z. & Butler, W. T. The Marfan syndrome: A deficiency in chemically stable collagen cross-links. N. Engl. J. Med.305(17), 988–991 (1981). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
