

Quantitative characterization of cell niches in spatially resolved omics data
- PMID: 40102688
- PMCID: PMC11985353
- DOI: 10.1038/s41588-025-02120-6
Quantitative characterization of cell niches in spatially resolved omics data
Abstract
Spatial omics enable the characterization of colocalized cell communities that coordinate specific functions within tissues. These communities, or niches, are shaped by interactions between neighboring cells, yet existing computational methods rarely leverage such interactions for their identification and characterization. To address this gap, here we introduce NicheCompass, a graph deep-learning method that models cellular communication to learn interpretable cell embeddings that encode signaling events, enabling the identification of niches and their underlying processes. Unlike existing methods, NicheCompass quantitatively characterizes niches based on communication pathways and consistently outperforms alternatives. We show its versatility by mapping tissue architecture during mouse embryonic development and delineating tumor niches in human cancers, including a spatial reference mapping application. Finally, we extend its capabilities to spatial multi-omics, demonstrate cross-technology integration with datasets from different sequencing platforms and construct a whole mouse brain spatial atlas comprising 8.4 million cells, highlighting NicheCompass' scalability. Overall, NicheCompass provides a scalable framework for identifying and analyzing niches through signaling events.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: S.B. is a part-time employee at Avanade Deutschland. M.L. owns interests in Relation Therapeutics and is a scientific cofounder and part-time employee at AIVIVO. F.J.T. consults for Immunai Inc., Singularity Bio B.V., CytoReason Ltd. and Omniscope and has an ownership interest in Dermagnostix GmbH and Cellarity. As of 1 February, 2025, C.T-L. is an employee at Cellzome GmbH/GSK. His contributions were done while being at the University of Würzburg. The other authors declare no competing interests.
Figures






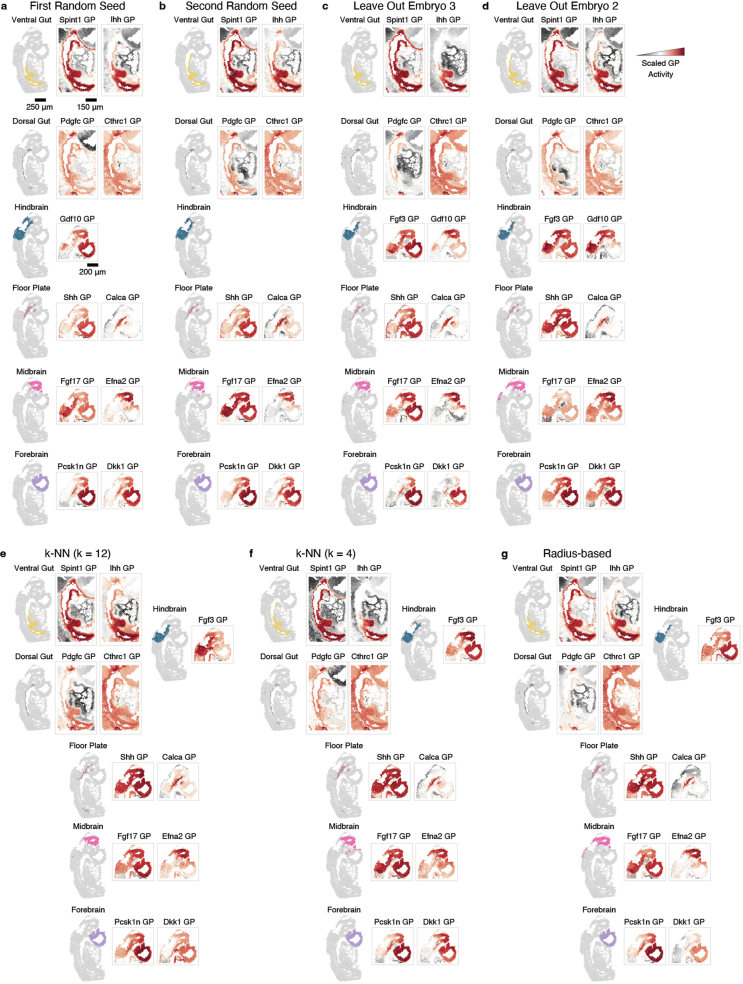
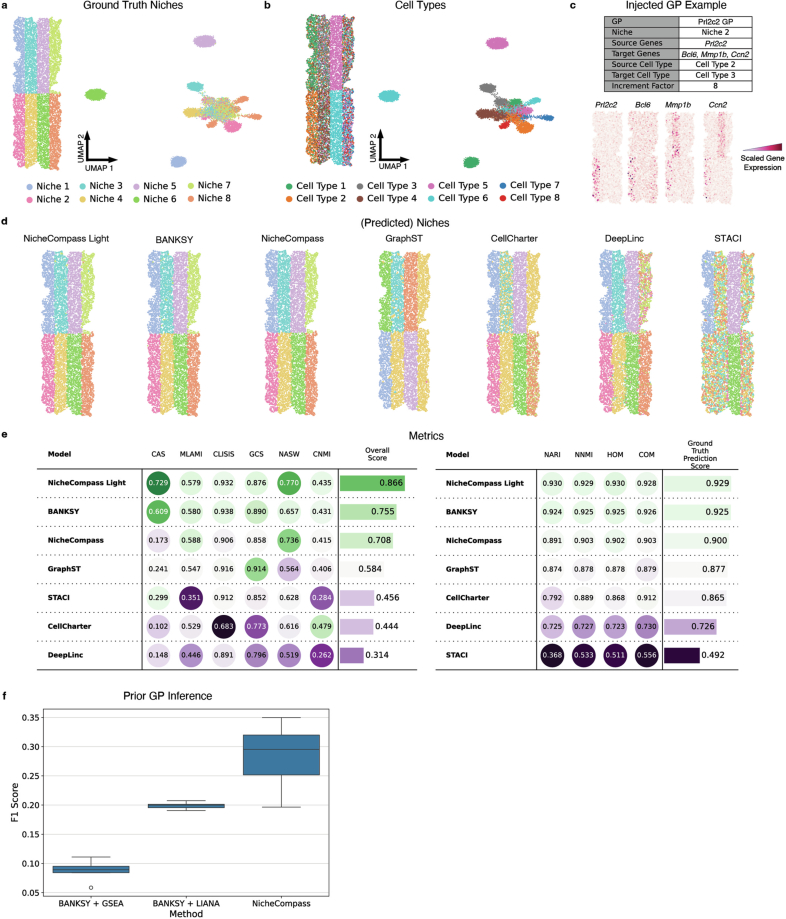







References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
