

Clinical and biochemical characteristics of patients with ornithine transcarbamylase deficiency and in silico analysis of OTC gene
- PMID: 40102887
- PMCID: PMC11916849
- DOI: 10.1186/s13023-025-03624-4
Clinical and biochemical characteristics of patients with ornithine transcarbamylase deficiency and in silico analysis of OTC gene
Abstract
Background: This study seeks to elucidate the clinical and biochemical features of Ornithine transcarbamylase deficiency (OTCD), a pleomorphic congenital hyperammonemia disorder with a non-specific clinical phenotype. Additionally, the research aims to analyze the mutation spectrum of the OTC gene and its potential association with phenotype, as well as to perform an in silico analysis of novel OTC variants to elucidate their structure-function relationship.
Methods: In this study, we conducted a retrospective analysis of the clinical and biochemical features of 12 patients with OTCD and examined their metabolite profiles. Additionally, we reviewed existing literature to explore the range of mutations in the OTC gene and their possible associations with phenotypic outcomes. Furthermore, we employed the high ambiguity-driven protein-protein docking (HADDOCK) algorithm and protein-ligand interaction profiler (PLIP) to predict the pathogenicity of these mutations and elucidate the underlying mechanisms of pathogenesis in novel variants of the OTC gene.
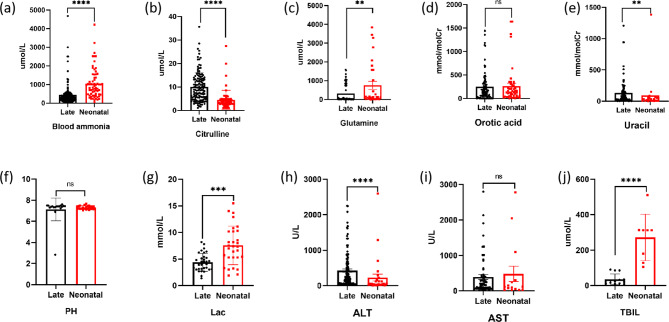
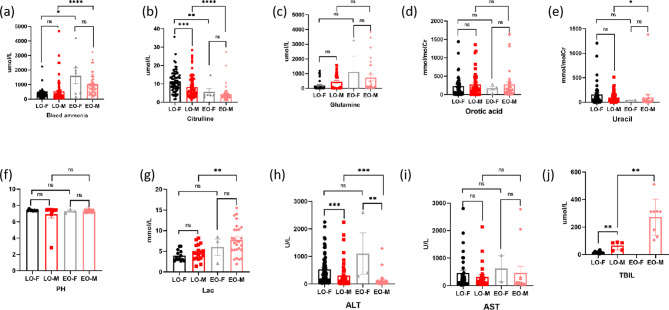
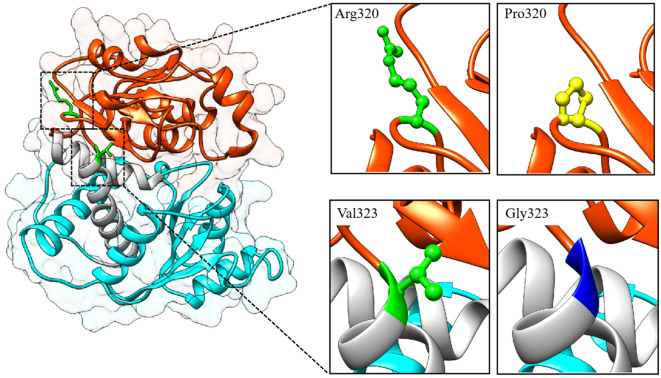
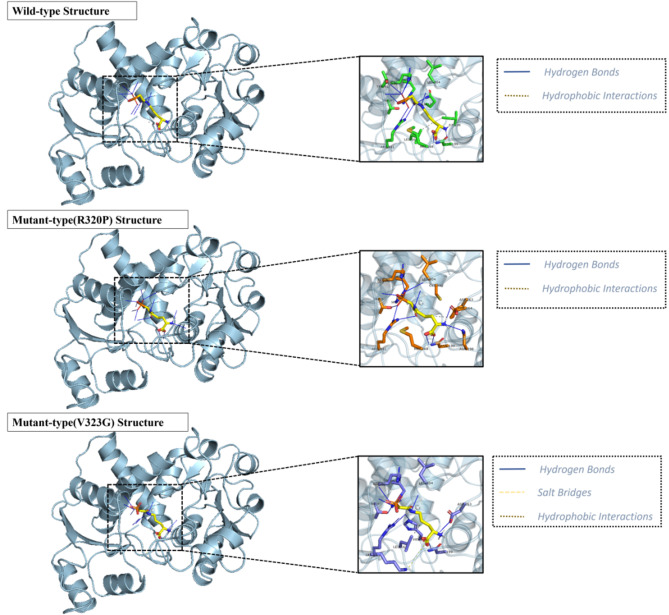
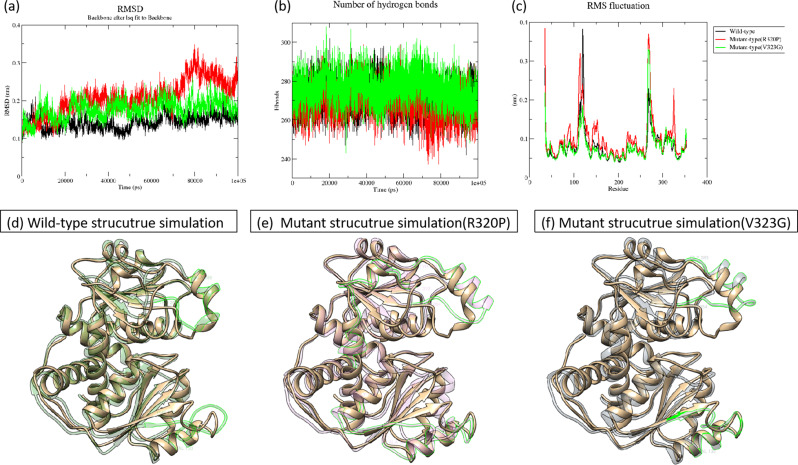
Results: Nine cases, all of which were male, presented with early onset, while two cases, all of which were female, exhibited late onset. Additionally, one male case was asymptomatic. The ages of the patients at the time of diagnosis ranged from 1 day to 12 years. Peak plasma ammonia levels were found to be higher in patients with early onset compared to those with late onset. Molecular analyses identified a total of 12 different mutations, including two novel mutations (V323G and R320P). In silico analysis indicated a potential difference in affinity between wild-type and mutant OTCase, with V323G and R320P mutations leading to a decreased binding ability of OTCase to the substrate, potentially disrupting its function.
Conclusion: This study broadened the genetic variation spectrum of OTCD and provided substantial evidence for genetic counselling to affected families. Additionally, we elucidated variant data of OTC in Chinese patients through comprehensive literature review. Given the ongoing uncertainty surrounding the genotype-phenotype correlation of OTCD, the results of our in silico analysis can contribute to a deeper understanding of this complex, rare, and severe genetic disorder.
Keywords: OTC; Gene variants; In Silico snalysis; Ornithine transcarbamylase defciency.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Clinical data of 12 OTCD patients were collected in the pediatric inpatient or outpatient department of the Sixth Affiliated Hospital, Sun Yat-sen University, and collected with ethical approval(2024ZSLYEC-393, 2023-063). Competing interests: The authors declare that they have no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
