

Gene-specific transcript buffering revealed by perturbation of coactivator complexes
- PMID: 40106549
- PMCID: PMC11922027
- DOI: 10.1126/sciadv.adr1492
Gene-specific transcript buffering revealed by perturbation of coactivator complexes
Abstract
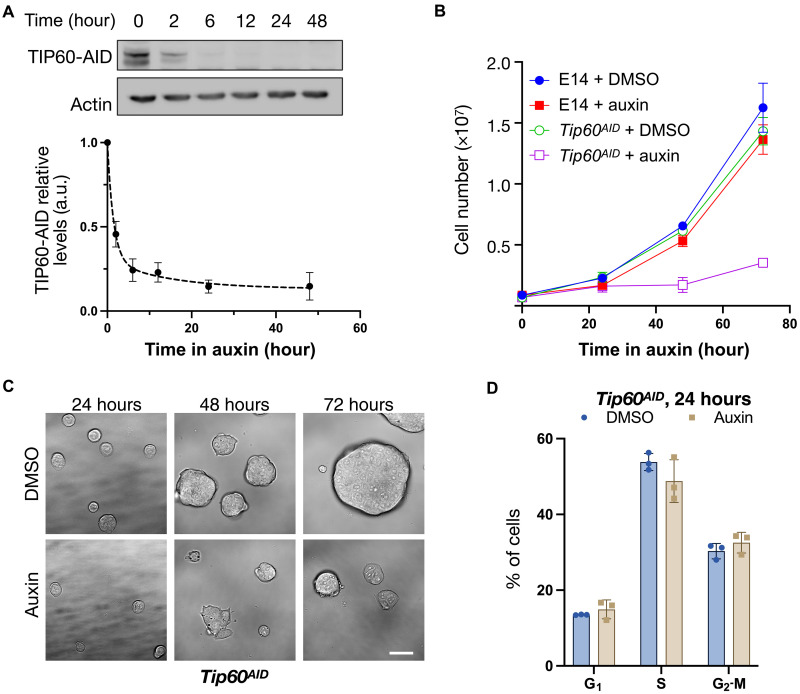
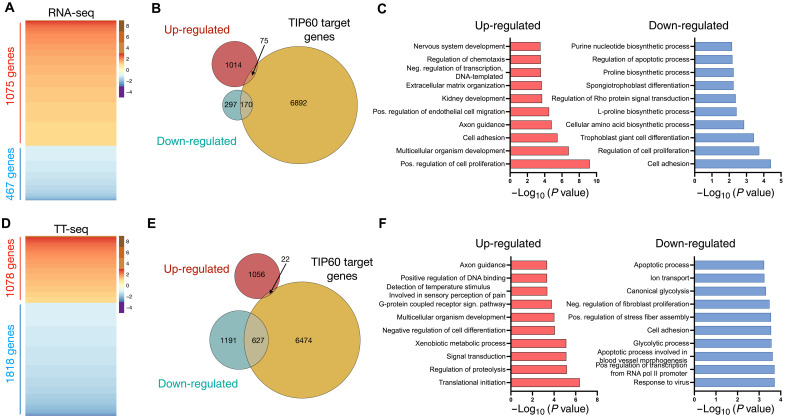
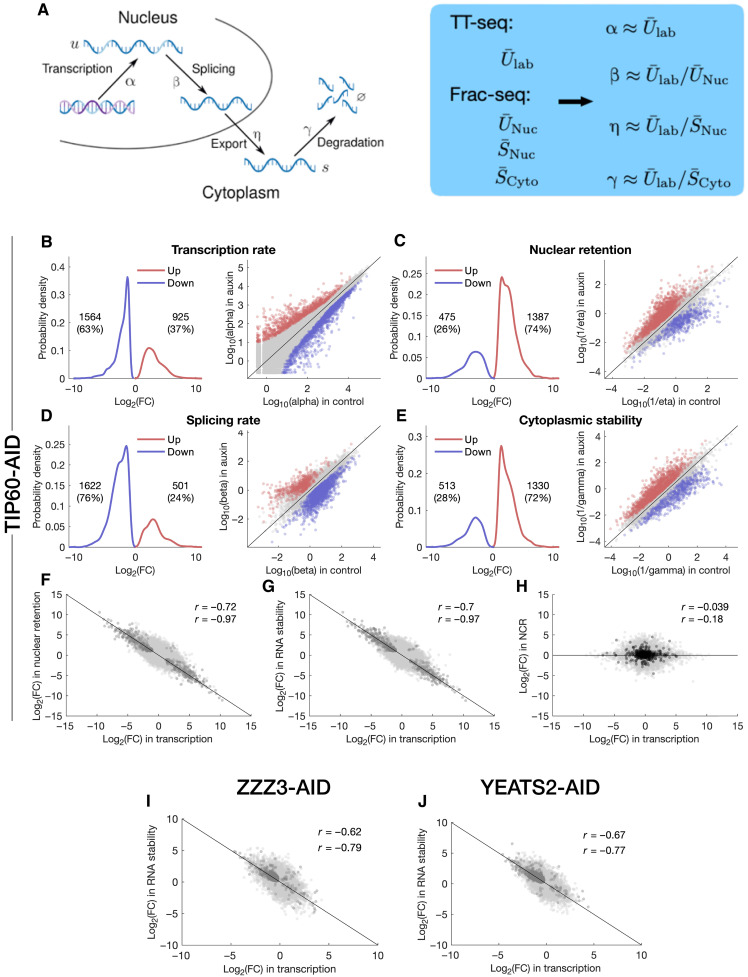
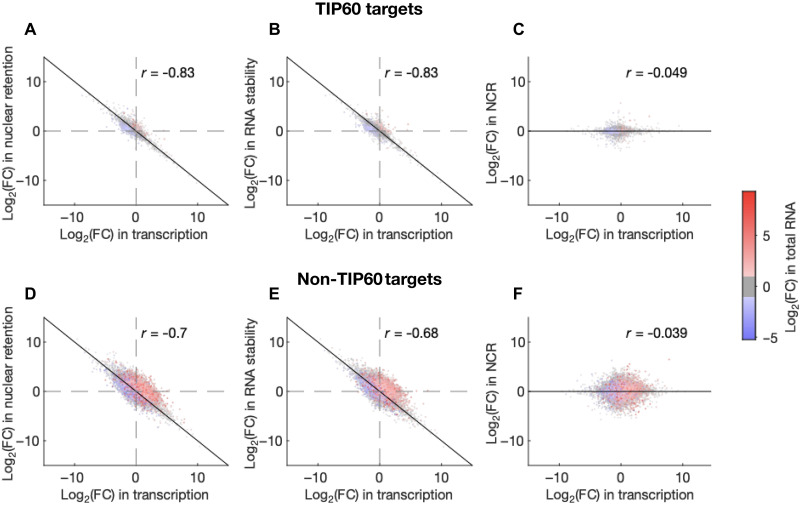
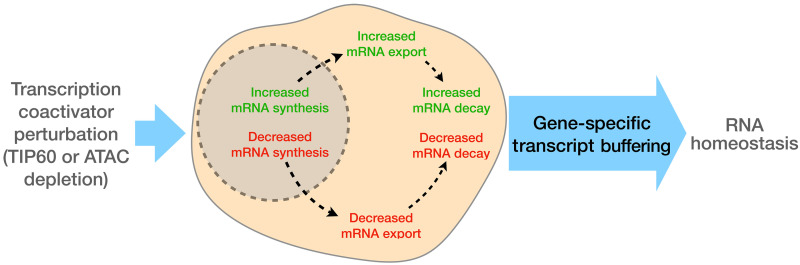
Transcript buffering entails reciprocal modulation of mRNA synthesis and degradation to maintain stable RNA levels under varying cellular conditions. Current models depict a global connection between mRNA synthesis and degradation, but underlying mechanisms remain unclear. Here, we show that changes in RNA metabolism following depletion of TIP60/KAT5, the acetyltransferase subunit of the NuA4 transcriptional coactivator complex, reveal that transcript buffering occurs at a gene-specific level. By combining RNA sequencing of nuclear, cytoplasmic, and newly synthesized transcript fractions with biophysical modeling in mouse embryonic stem cells, we demonstrate that transcriptional changes caused by TIP60 depletion are offset by corresponding changes in RNA nuclear export and cytoplasmic stability, indicating gene-specific buffering. Disruption of the unrelated ATAC coactivator complex also causes gene-specific transcript buffering. We propose that cells dynamically adjust RNA splicing, export, and degradation in response to individual RNA synthesis alterations, thereby sustaining cellular homeostasis.
Figures
Update of
-
Gene-specific RNA homeostasis revealed by perturbation of coactivator complexes.bioRxiv [Preprint]. 2024 May 7:2024.01.30.577960. doi: 10.1101/2024.01.30.577960. bioRxiv. 2024. Update in: Sci Adv. 2025 Mar 21;11(12):eadr1492. doi: 10.1126/sciadv.adr1492. PMID: 38352321 Free PMC article. Updated. Preprint.
References
-
- Berry S., Müller M., Rai A., Pelkmans L., Feedback from nuclear RNA on transcription promotes robust RNA concentration homeostasis in human cells. Cell Syst. 13, 454–470.e15 (2022). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
