

VDAC2 loss elicits tumour destruction and inflammation for cancer therapy
- PMID: 40108474
- PMCID: PMC12018455
- DOI: 10.1038/s41586-025-08732-6
VDAC2 loss elicits tumour destruction and inflammation for cancer therapy
Abstract
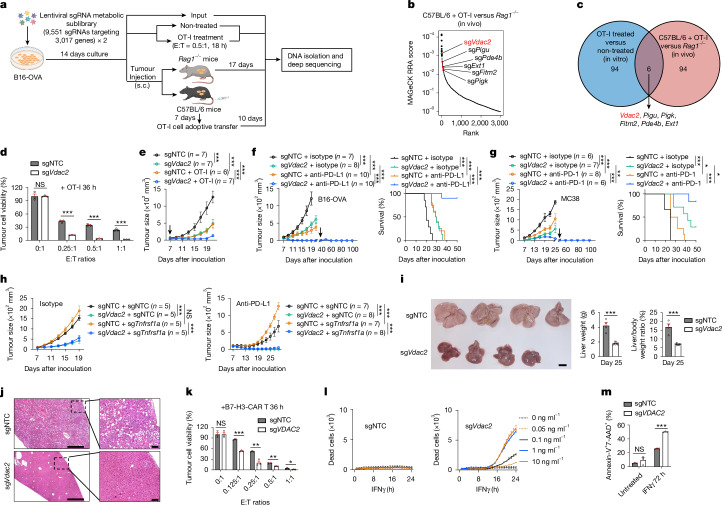
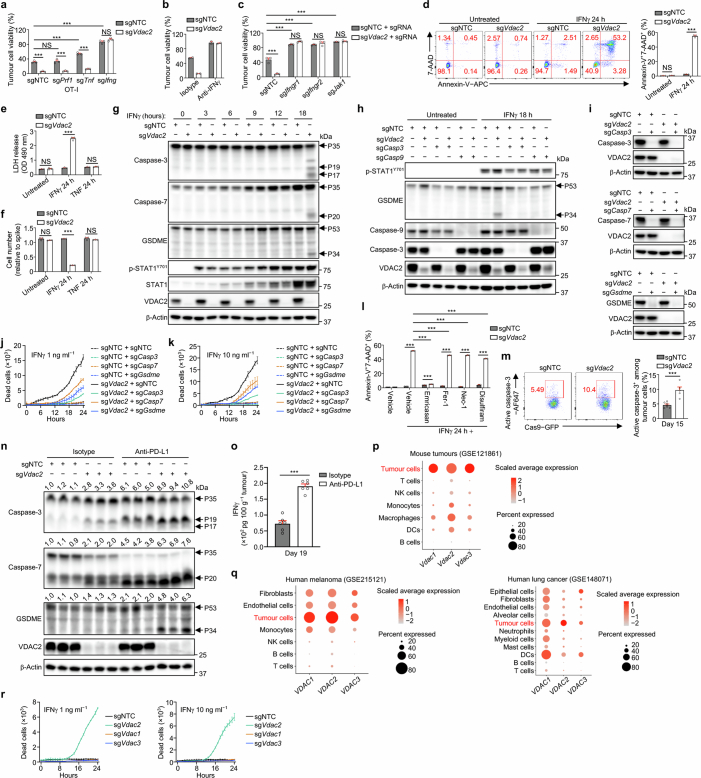
Tumour cells often evade immune pressure exerted by CD8+ T cells or immunotherapies through mechanisms that are largely unclear1,2. Here, using complementary in vivo and in vitro CRISPR-Cas9 genetic screens to target metabolic factors, we established voltage-dependent anion channel 2 (VDAC2) as an immune signal-dependent checkpoint that curtails interferon-γ (IFNγ)-mediated tumour destruction and inflammatory reprogramming of the tumour microenvironment. Targeting VDAC2 in tumour cells enabled IFNγ-induced cell death and cGAS-STING activation, and markedly improved anti-tumour effects and immunotherapeutic responses. Using a genome-scale genetic interaction screen, we identified BAK as the mediator of VDAC2-deficiency-induced effects. Mechanistically, IFNγ stimulation increased BIM, BID and BAK expression, with VDAC2 deficiency eliciting uncontrolled IFNγ-induced BAK activation and mitochondrial damage. Consequently, mitochondrial DNA was aberrantly released into the cytosol and triggered robust activation of cGAS-STING signalling and type I IFN response. Importantly, co-deletion of STING signalling components dampened the therapeutic effects of VDAC2 depletion in tumour cells, suggesting that targeting VDAC2 integrates CD8+ T cell- and IFNγ-mediated adaptive immunity with a tumour-intrinsic innate immune-like response. Together, our findings reveal VDAC2 as a dual-action target to overcome tumour immune evasion and establish the importance of coordinately destructing and inflaming tumours to enable efficacious cancer immunotherapy.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: S.G. is a member of the Scientific Advisory Board of Be Biopharma and CARGO and the Data and Safety Monitoring Board (DSMB) of Immatics, and is a co-inventor on patents/patent applications in the fields of immunotherapy and gene therapy. H.C. consults for Kumquat Biosciences and TCura Bioscience, and is a co-inventor on patents/patent applications in the field of immunotherapy. The remaining authors declare no competing interests.
Figures












References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
