

FGR Src family kinase causes signaling and phenotypic shift mimicking retinoic acid-induced differentiation of leukemic cells
- PMID: 40116400
- PMCID: PMC11927794
- DOI: 10.18632/oncotarget.28705
FGR Src family kinase causes signaling and phenotypic shift mimicking retinoic acid-induced differentiation of leukemic cells
Abstract
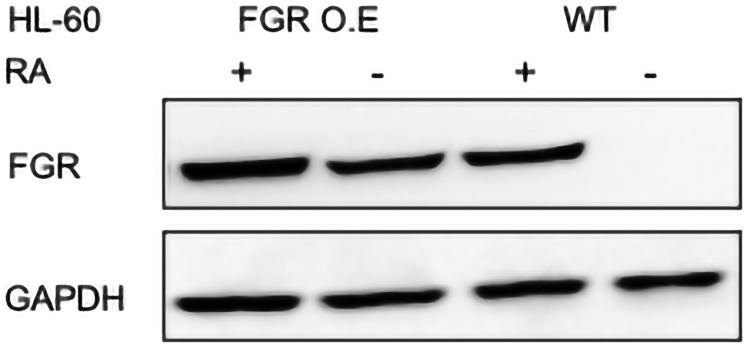
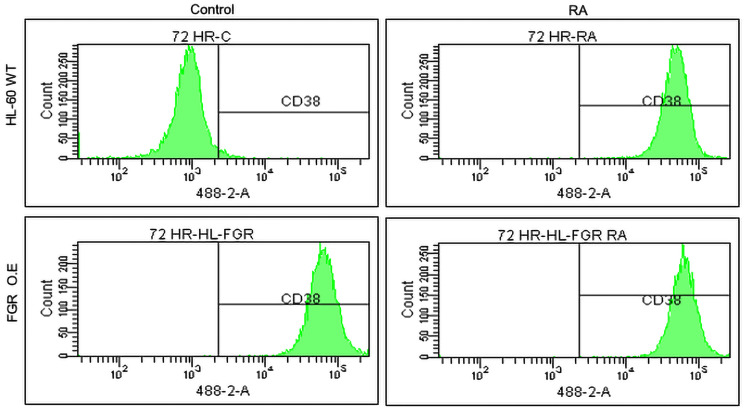
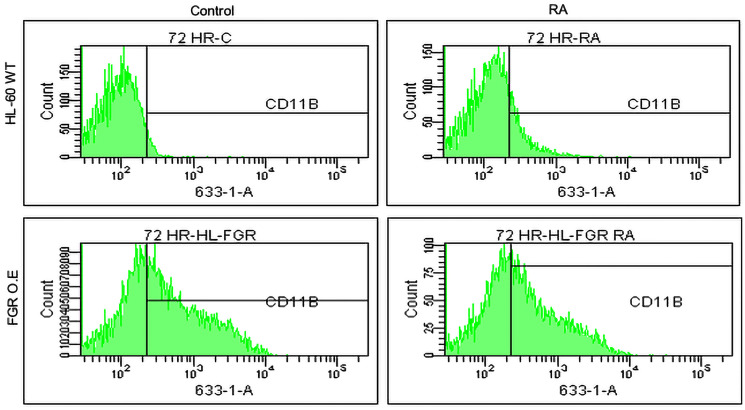
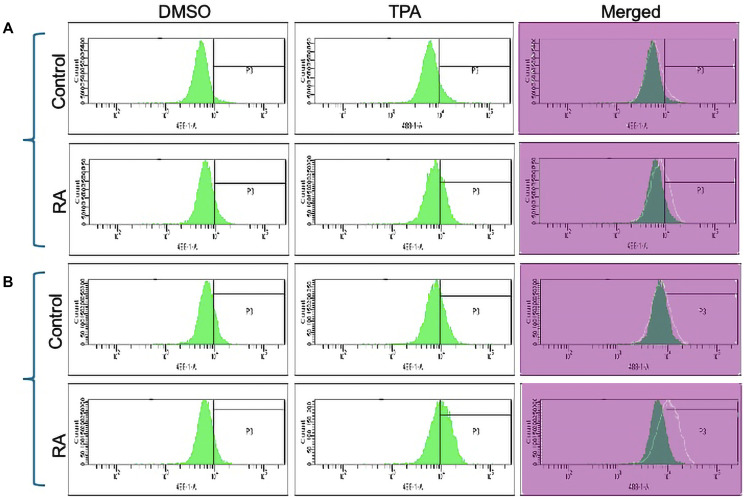
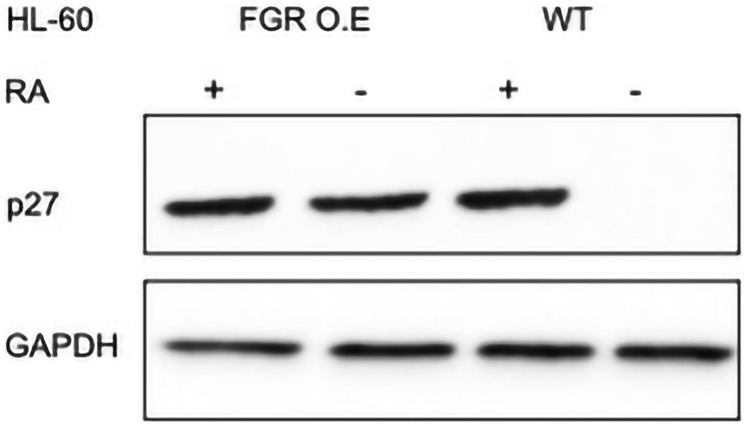
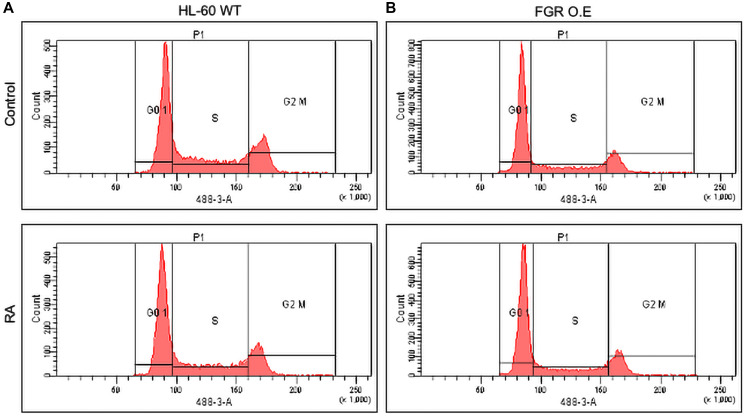
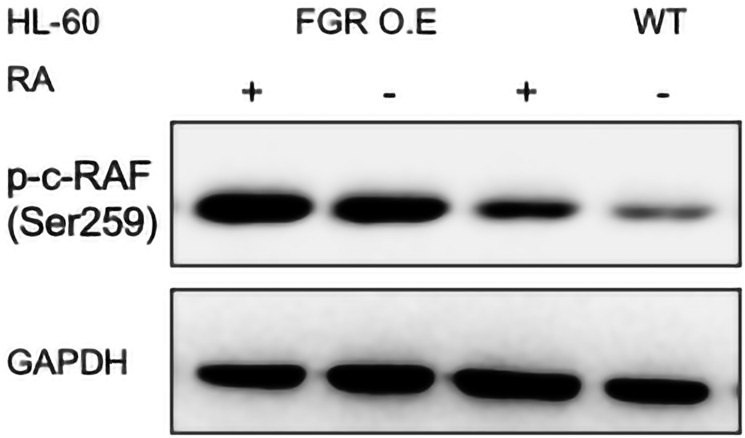
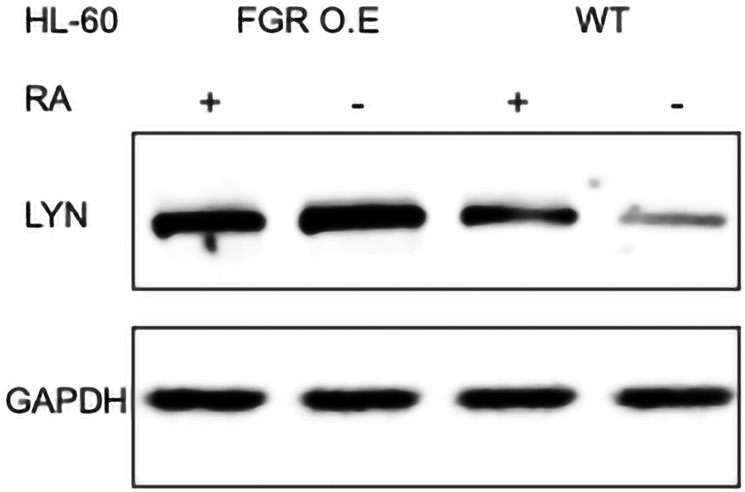
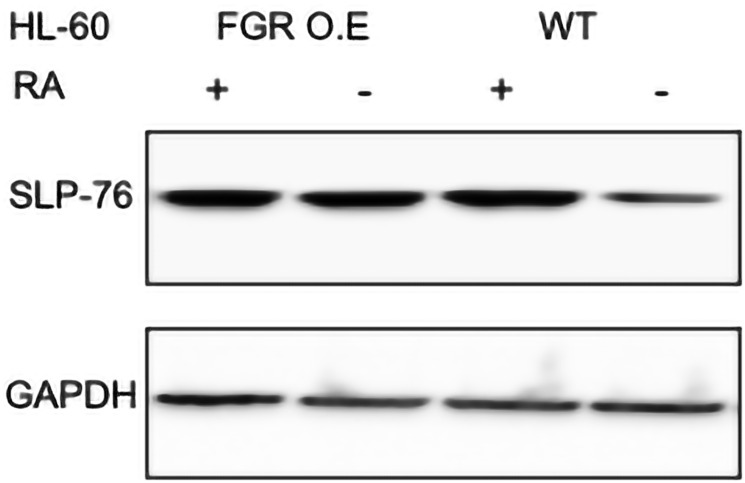
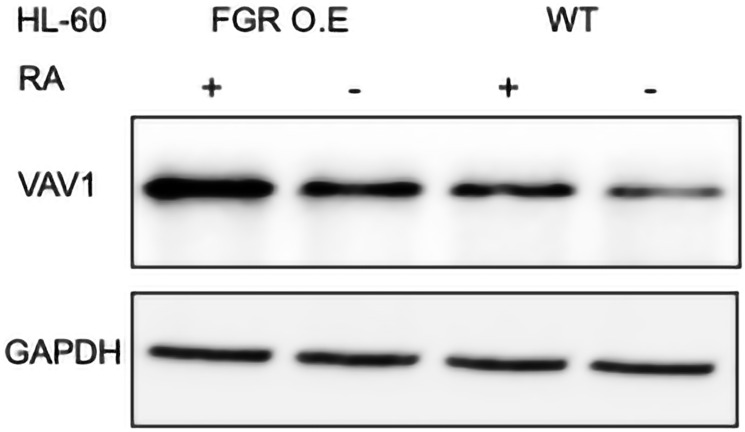
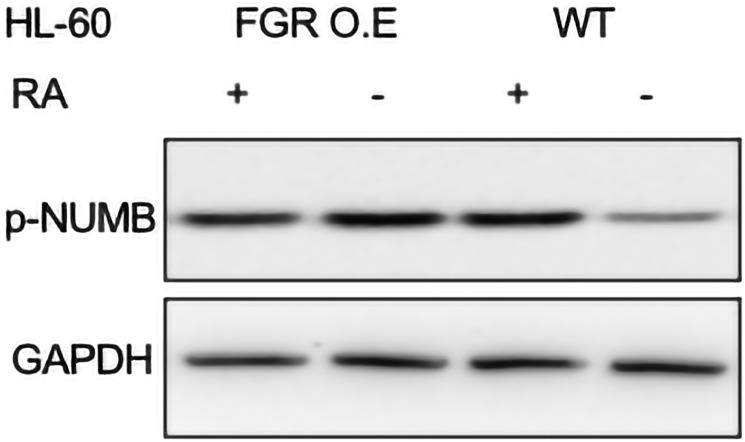
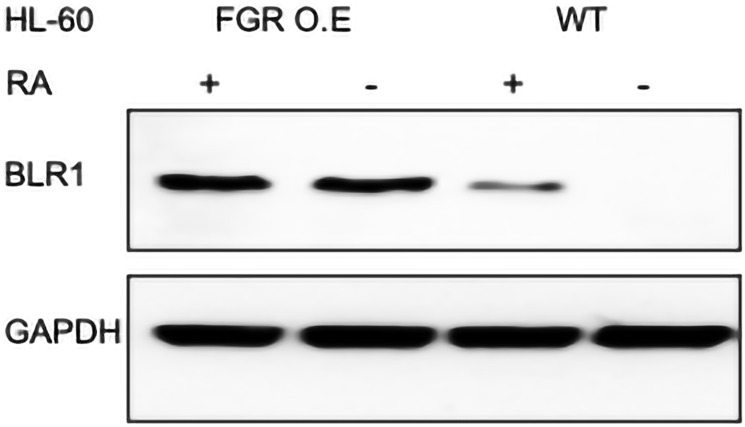
Retinoic acid (RA), an embryonic morphogen, is used in cancer differentiation therapy, causing extensive gene expression changes leading to cell differentiation. This study reveals that the expression of the Src-family kinase (SFK), FGR, alone can induce cell differentiation similar to RA. Traditionally, RA's mechanism involves transcriptional activation via RAR/RXR(Retinoic Acid Receptor/Retinoid X Receptor) nuclear receptors. In the HL-60 human myelo-monocytic leukemia model, an actively proliferating phenotypically immature, lineage bipotent NCI-60 cell line. RA promotes myeloid lineage selection and maturation with G1/0 growth inhibition. This study finds that FGR expression alone is sufficient to induce differentiation, marked by CD38, CD11b, ROS, and p27(kip1) expression, characteristic of mature myeloid cells. To understand the mechanism, signaling attributes promoting RA-induced differentiation were analyzed. RA induces FGR expression, which activates a novel cytosolic macromolecular signaling complex(signalsome) driving differentiation. RA increases the abundance, associations, and phosphorylation of signalsome components, including RAF, LYN, FGR, SLP-76, and CBL, which appear as nodes in the signalsome. These traditionally cytosolic signaling molecules go into the nucleus. RAF complexes with a retinoic acid-response element (RARE) in the blr1 gene promoter, where the induced BLR1 expression is essential for RA-induced differentiation. We find now that FGR expression mimics RA's enhancement of signalsome nodes, RAF expression, and phosphorylation, leading to BLR1 expression. Notably, FGR induces the expression of genes targeted by RAR/RXR, such as cd38 and blr1, even without RA. Thus, FGR triggers signaling events and phenotypic shifts characteristic of RA. This finding represents a paradigm shift, given FGR's historical role as a pro-proliferation oncogene.
Keywords: FGR Src-family-kinase; cancer biology and cell cycle regulation; cancer differentiation-therapy; leukemia; retinoic acid(RA).
Conflict of interest statement
Authors have no conflicts of interest to declare.
Figures

Similar articles
-
Roscovitine enhances All-trans retinoic acid (ATRA)-induced leukemia cell differentiation: Novel effects on signaling molecules for a putative Cdk2 inhibitor.Cell Signal. 2020 Jul;71:109555. doi: 10.1016/j.cellsig.2020.109555. Epub 2020 Feb 4. Cell Signal. 2020. PMID: 32032659
-
6-Formylindolo(3,2-b)Carbazole (FICZ) Modulates the Signalsome Responsible for RA-Induced Differentiation of HL-60 Myeloblastic Leukemia Cells.PLoS One. 2015 Aug 19;10(8):e0135668. doi: 10.1371/journal.pone.0135668. eCollection 2015. PLoS One. 2015. PMID: 26287494 Free PMC article.
-
The Src-family kinase inhibitor PP2 rescues inducible differentiation events in emergent retinoic acid-resistant myeloblastic leukemia cells.PLoS One. 2013;8(3):e58621. doi: 10.1371/journal.pone.0058621. Epub 2013 Mar 15. PLoS One. 2013. PMID: 23554907 Free PMC article.
-
GW5074 and PP2 kinase inhibitors implicate nontraditional c-Raf and Lyn function as drivers of retinoic acid-induced maturation.Cell Signal. 2015 Aug;27(8):1666-75. doi: 10.1016/j.cellsig.2015.03.014. Epub 2015 Mar 26. Cell Signal. 2015. PMID: 25817574 Free PMC article.
-
Mechanisms of all-trans retinoic acid-induced differentiation of acute promyelocytic leukemia cells.J Biosci. 2000 Sep;25(3):275-84. doi: 10.1007/BF02703936. J Biosci. 2000. PMID: 11022230 Review.
Cited by
-
New Fluorescent Synthetic Retinoids as Potential RAR Agonists with Anticancer, Molecular Docking and ADME Assessments.J Fluoresc. 2025 May 23. doi: 10.1007/s10895-025-04343-6. Online ahead of print. J Fluoresc. 2025. PMID: 40410548
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
