

Long-read sequencing identifies copy-specific markers of SMN gene conversion in spinal muscular atrophy
- PMID: 40119448
- PMCID: PMC11927269
- DOI: 10.1186/s13073-025-01448-2
Long-read sequencing identifies copy-specific markers of SMN gene conversion in spinal muscular atrophy
Abstract
Background: The complex 2 Mb survival motor neuron (SMN) locus on chromosome 5q13, including the spinal muscular atrophy (SMA)-causing gene SMN1 and modifier SMN2, remains incompletely resolved due to numerous segmental duplications. Variation in SMN2 copy number, presumably influenced by SMN1 to SMN2 gene conversion, affects disease severity, though SMN2 copy number alone has insufficient prognostic value due to limited genotype-phenotype correlations. With advancements in newborn screening and SMN-targeted therapies, identifying genetic markers to predict disease progression and treatment response is crucial. Progress has thus far been limited by methodological constraints.
Methods: To address this, we developed HapSMA, a method to perform polyploid phasing of the SMN locus to enable copy-specific analysis of SMN and its surrounding genes. We used HapSMA on publicly available Oxford Nanopore Technologies (ONT) sequencing data of 29 healthy controls and performed long-read, targeted ONT sequencing of the SMN locus of 31 patients with SMA.
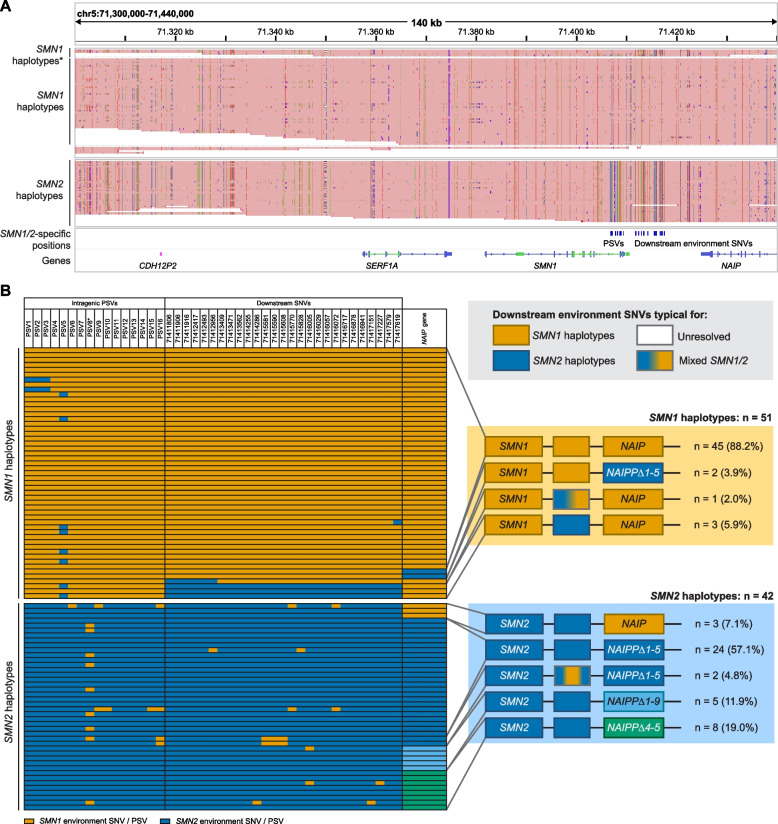
Results: In healthy controls, we identified single nucleotide variants (SNVs) specific to SMN1 and SMN2 haplotypes that could serve as gene conversion markers. Broad phasing including the NAIP gene allowed for a more complete view of SMN locus variation. Genetic variation in SMN2 haplotypes was larger in SMA patients. Forty-two percent of SMN2 haplotypes of SMA patients showed varying SMN1 to SMN2 gene conversion breakpoints, serving as direct evidence of gene conversion as a common genetic characteristic in SMA and highlighting the importance of inclusion of SMA patients when investigating the SMN locus.
Conclusions: Our findings illustrate that both methodological advances and the analysis of patient samples are required to advance our understanding of complex genetic loci and address critical clinical challenges.
Keywords: Dark genomic regions; Gene conversion; Long-read sequencing; Segmental duplications; Spinal muscular atrophy.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: The study protocol (09307/NL29692.041.09) was approved by the Medical Ethical Committee of the University Medical Center Utrecht and registered at the Dutch registry for clinical studies and trials [24]. Written informed consent was obtained from all adult patients, and from patients and/or parents additionally in the case of children younger than 18 years old. Consent for publication: Not applicable. Competing interests: JHV reports to have sponsored research agreements with Biogen and Astra Zeneca. The remaining authors declare that they do not have any competing interests.
Figures


References
-
- Schmutz J, Martin J, Terry A, Couronne O, Grimwood J, Lowry S, et al. The DNA sequence and comparative analysis of human chromosome 5. Nature. 2004;431:268–74. - PubMed
-
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–65. - PubMed
-
- Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AHM, et al. A Single Nucleotide Difference That Alters Splicing Patterns Distinguishes the SMA Gene SMN1 From the Copy Gene SMN2. Hum Mol Genet. 1999;8:1177–83. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
