

Integrative genomics sheds light on the immunogenetics of tuberculosis in cattle
- PMID: 40128580
- PMCID: PMC11933339
- DOI: 10.1038/s42003-025-07846-x
Integrative genomics sheds light on the immunogenetics of tuberculosis in cattle
Abstract
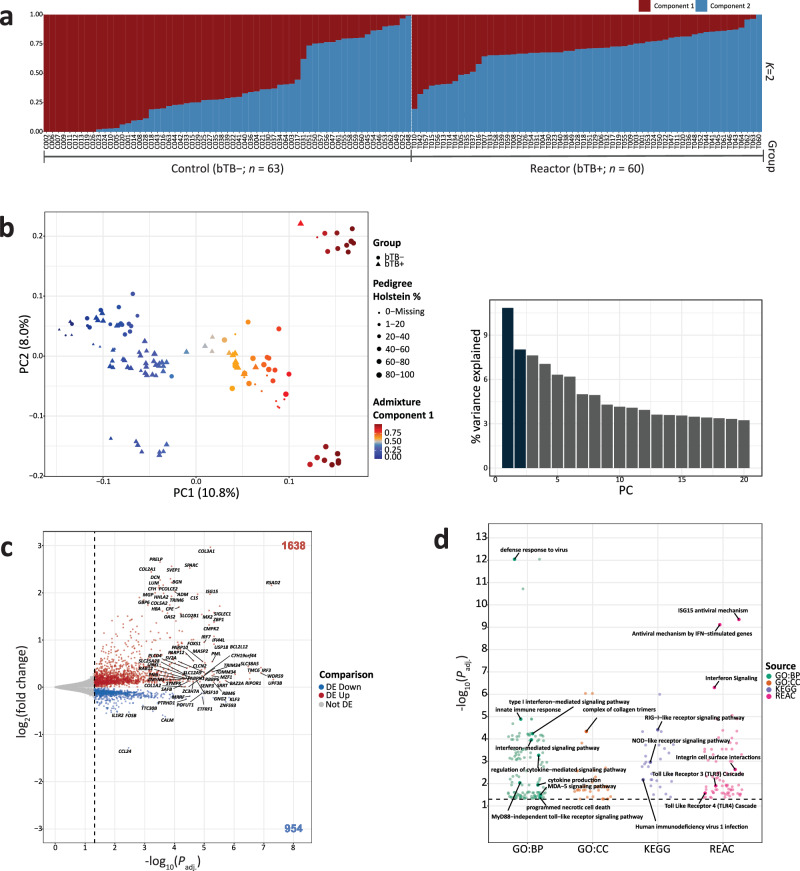
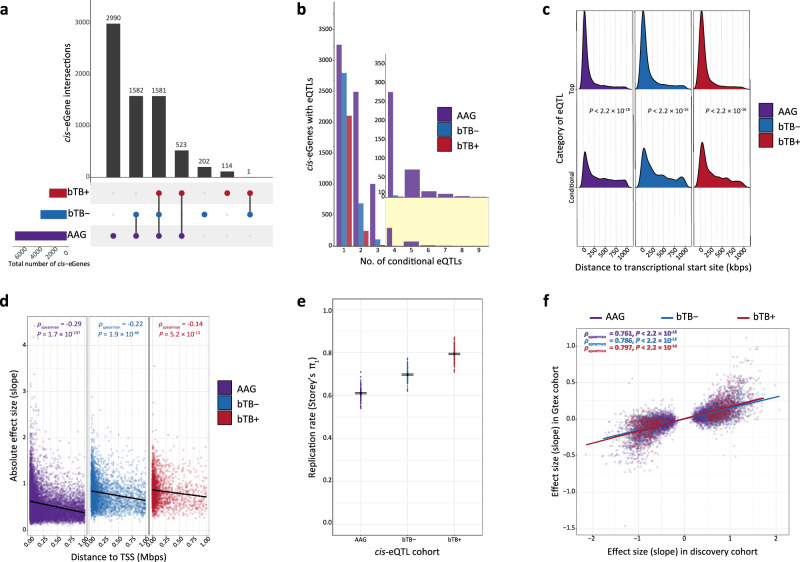
Mycobacterium bovis causes bovine tuberculosis (bTB), an infectious disease of cattle that represents a zoonotic threat to humans. Research has shown that the peripheral blood (PB) transcriptome is perturbed during bTB disease but the genomic architecture underpinning this transcriptional response remains poorly understood. Here, we analyse PB transcriptomics data from 63 control and 60 confirmed M. bovis-infected animals and detect 2592 differently expressed genes perturbing multiple immune response pathways. Leveraging imputed genome-wide SNP data, we characterise thousands of cis-expression quantitative trait loci (eQTLs) and show that the PB transcriptome is substantially impacted by intrapopulation genomic variation during M. bovis infection. Integrating our cis-eQTL data with bTB susceptibility GWAS summary statistics, we perform a transcriptome-wide association study and identify 115 functionally relevant genes (including RGS10, GBP4, TREML2, and RELT) and provide important new omics data for understanding the host response to mycobacterial infections that cause tuberculosis in mammals.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures



References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
