

Elementary methods provide more replicable results in microbial differential abundance analysis
- PMID: 40135504
- PMCID: PMC11937625
- DOI: 10.1093/bib/bbaf130
Elementary methods provide more replicable results in microbial differential abundance analysis
Abstract
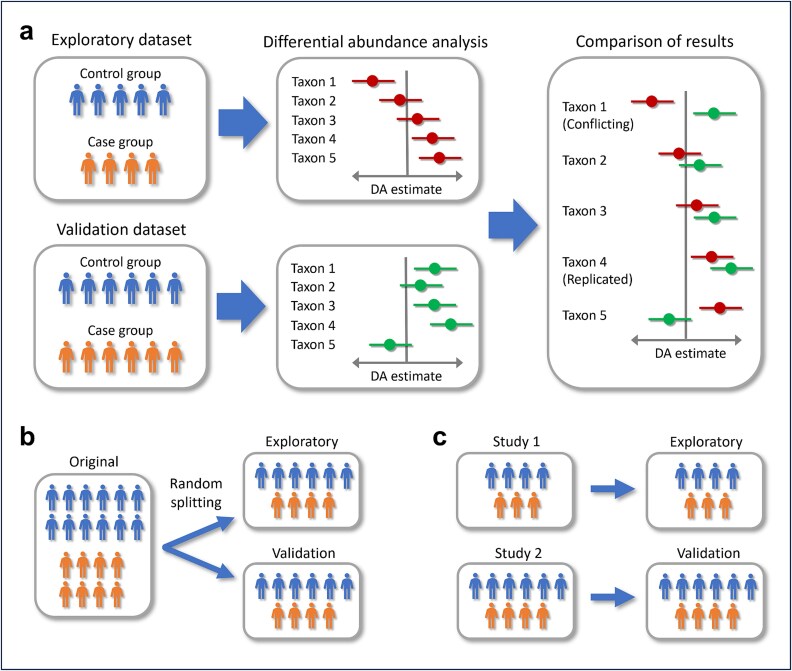
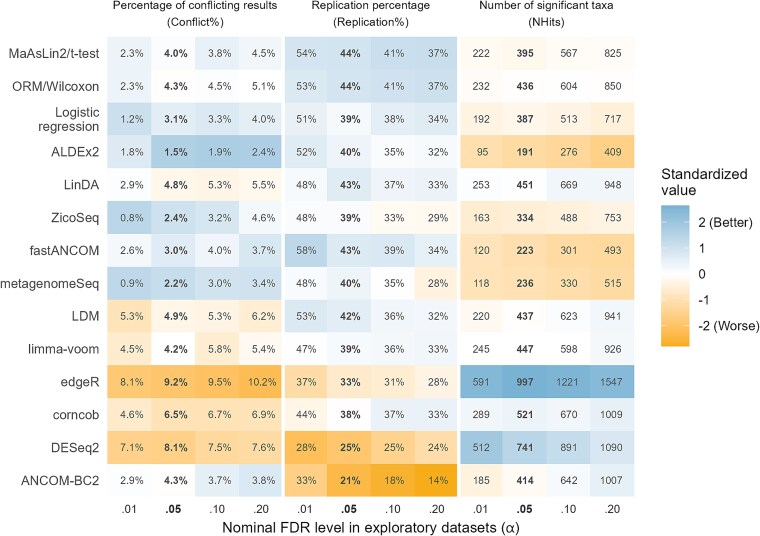
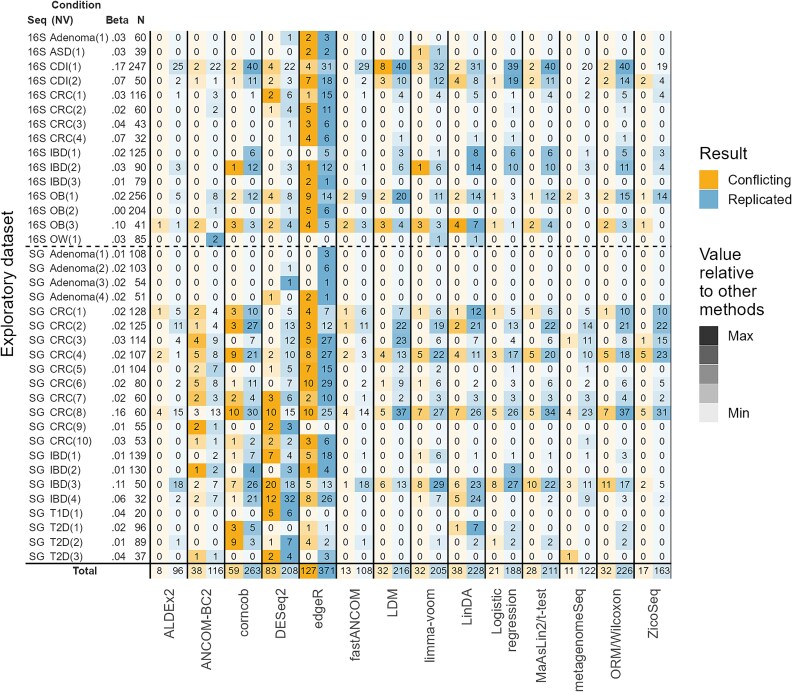
Differential abundance analysis (DAA) is a key component of microbiome studies. Although dozens of methods exist, there is currently no consensus on the preferred methods. While the correctness of results in DAA is an ambiguous concept and cannot be fully evaluated without setting the ground truth and employing simulated data, we argue that a well-performing method should be effective in producing highly reproducible results. We compared the performance of 14 DAA methods by employing datasets from 53 taxonomic profiling studies based on 16S rRNA gene or shotgun metagenomic sequencing. For each method, we examined how the results replicated between random partitions of each dataset and between datasets from separate studies. While certain methods showed good consistency, some widely used methods were observed to produce a substantial number of conflicting findings. Overall, when considering consistency together with sensitivity, the best performance was attained by analyzing relative abundances with a nonparametric method (Wilcoxon test or ordinal regression model) or linear regression/t-test. Moreover, a comparable performance was obtained by analyzing presence/absence of taxa with logistic regression.
Keywords: benchmarking; differential abundance analysis; microbiome; replicability.
© The Author(s) 2025. Published by Oxford University Press.
Figures


Similar articles
-
Benchmarking Differential Abundance Tests for 16S microbiome sequencing data using simulated data based on experimental templates.PLoS One. 2025 May 19;20(5):e0321452. doi: 10.1371/journal.pone.0321452. eCollection 2025. PLoS One. 2025. PMID: 40388544 Free PMC article.
-
Large-scale benchmarking reveals false discoveries and count transformation sensitivity in 16S rRNA gene amplicon data analysis methods used in microbiome studies.Microbiome. 2016 Nov 25;4(1):62. doi: 10.1186/s40168-016-0208-8. Microbiome. 2016. PMID: 27884206 Free PMC article.
-
Assessment of In Vitro and In Silico Protocols for Sequence-Based Characterization of the Human Vaginal Microbiome.mSphere. 2020 Nov 18;5(6):e00448-20. doi: 10.1128/mSphere.00448-20. mSphere. 2020. PMID: 33208514 Free PMC article.
-
Metatranscriptomics: A Tool for Clinical Metagenomics.OMICS. 2024 Aug;28(8):394-407. doi: 10.1089/omi.2024.0130. Epub 2024 Jul 19. OMICS. 2024. PMID: 39029911 Review.
-
Current challenges and best-practice protocols for microbiome analysis.Brief Bioinform. 2021 Jan 18;22(1):178-193. doi: 10.1093/bib/bbz155. Brief Bioinform. 2021. PMID: 31848574 Free PMC article. Review.
Cited by
-
MaAsLin 3: Refining and extending generalized multivariable linear models for meta-omic association discovery.bioRxiv [Preprint]. 2024 Dec 14:2024.12.13.628459. doi: 10.1101/2024.12.13.628459. bioRxiv. 2024. PMID: 39713460 Free PMC article. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
