

Does Early Diagnosis and Treatment Alter the Clinical Course of Wolman Disease? Divergent Trajectories in Two Siblings and a Consideration for Newborn Screening
- PMID: 40136632
- PMCID: PMC11943304
- DOI: 10.3390/ijns11010017
Does Early Diagnosis and Treatment Alter the Clinical Course of Wolman Disease? Divergent Trajectories in Two Siblings and a Consideration for Newborn Screening
Abstract
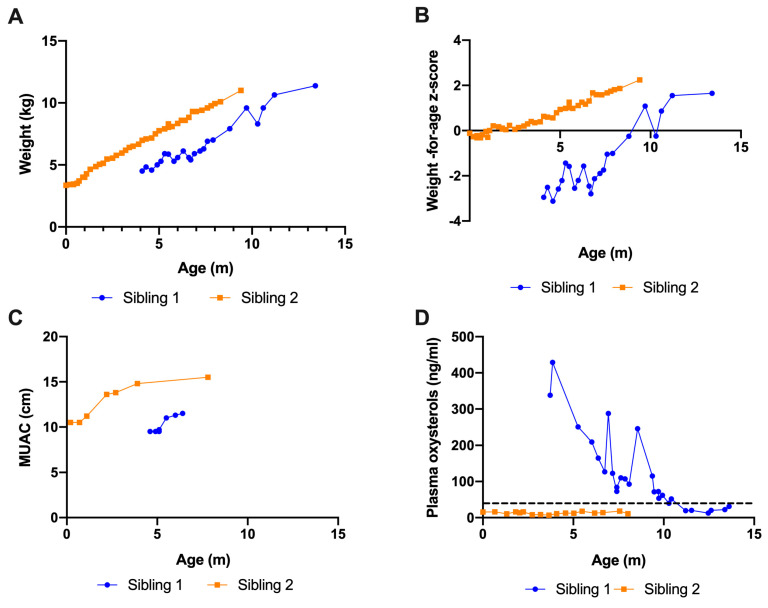
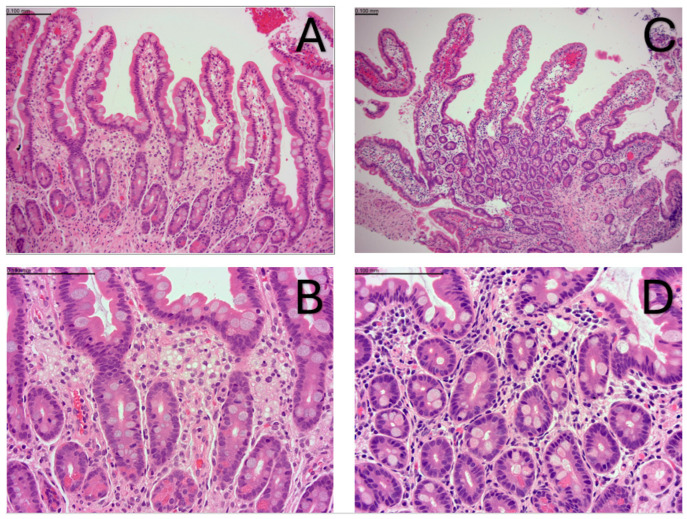
Wolman disease (WD) is a lethal disorder defined by the deficiency of the lysosomal acid lipase enzyme. Patients present with intestinal failure, malnutrition, and hepatosplenomegaly. Enzyme replacement therapy (ERT) with dietary substrate reduction (DSR) significantly improves survival. We sought to determine the outcomes of two siblings with WD treated after the onset of symptoms (sibling 1) and presymptomatic (sibling 2). A chart review was conducted on two siblings with WD treated with ERT and DSR at 4 months of age (sibling 1) and immediately after birth (sibling 2) to determine clinical outcomes based on survival, laboratory results, growth, dietary records, and gut biopsies. Sibling 1 presented with hepatosplenomegaly and liver dysfunction and developed hemophagocytic lymphohistiocytosis despite treatment. She received a bone marrow transplant at 8 months of age but died at 13 months. Sibling 2 is alive at 16 months of age with height, weight, and MUAC above the 95th centile, fully orally fed, with no gastrointestinal symptoms, normal liver function, and normal oxysterols. Sibling 2 duodenal biopsies show normal villus architecture with no foamy macrophage infiltration. Initiation of treatment prior to the onset of symptoms can prevent clinical manifestations and increase survival. The divergent trajectory in these siblings raises the question of WD's candidacy for newborn screening.
Keywords: Wolman disease; dietary substrate reduction; early diagnosis; early treatment; enzyme replacement therapy; newborn screening.
Conflict of interest statement
S.A.J., A.G. and F.W. provided consulting support to Alexion but not over the last 2 years. The other authors declare no conflicts of interest.
Figures
References
-
- Kohli R., Ratziu V., Fiel M.I., Waldmann E., Wilson D.P., Balwani M. Initial assessment and ongoing monitoring of lysosomal acid lipase deficiency in children and adults: Consensus recommendations from an international collaborative working group. Mol. Genet. Metab. 2020;129:59–66. doi: 10.1016/j.ymgme.2019.11.004. - DOI - PubMed
-
- Jones S.A., Valayannopoulos V., Schneider E., Eckert S., Banikazemi M., Bialer M., Cederbaum S., Chan A., Dhawan A., Di Rocco M., et al. Rapid progression and mortality of lysosomal acid lipase deficiency presenting in infants. Genet. Med. 2016;18:452–458. doi: 10.1038/gim.2015.108. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
