

Impact of a High-Fat Diet on the Gut Microbiome: A Comprehensive Study of Microbial and Metabolite Shifts During Obesity
- PMID: 40136712
- PMCID: PMC11940932
- DOI: 10.3390/cells14060463
Impact of a High-Fat Diet on the Gut Microbiome: A Comprehensive Study of Microbial and Metabolite Shifts During Obesity
Abstract
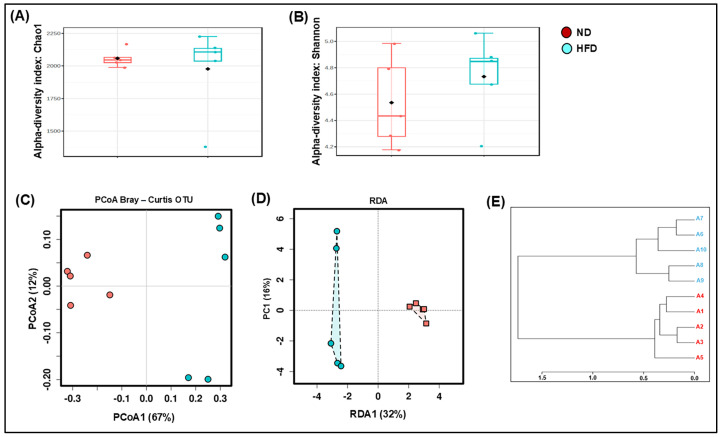
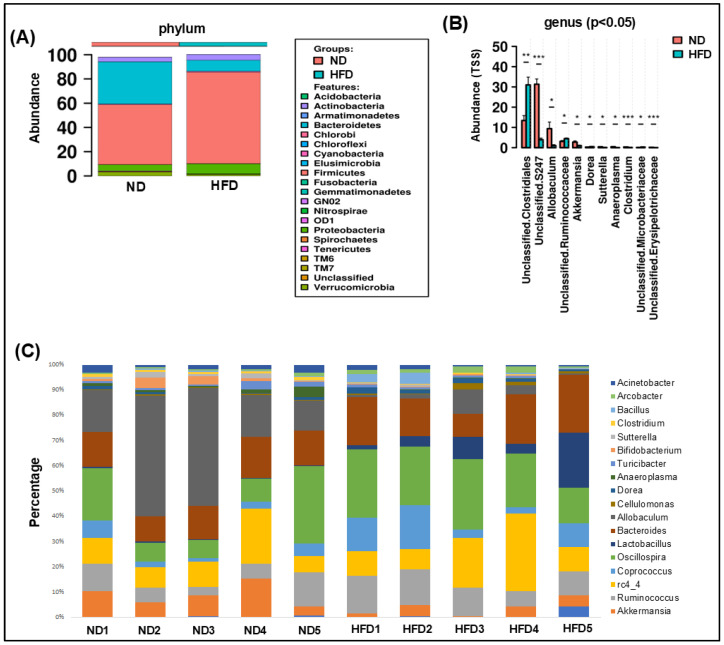
Over the last few decades, the prevalence of metabolic diseases such as obesity, diabetes, non-alcoholic fatty liver disease, hypertension, and hyperuricemia has surged, primarily due to high-fat diet (HFD). The pathologies of these metabolic diseases show disease-specific alterations in the composition and function of their gut microbiome. How HFD alters the microbiome and its metabolite to mediate adipose tissue (AT) inflammation and obesity is not well known. Thus, this study aimed to identify the changes in the gut microbiome and metabolomic signatures induced by an HFD to alter obesity. To explore the changes in the gut microbiota and metabolites, 16S rRNA gene amplicon sequencing and metabolomic analyses were performed after HFD and normal diet (ND) feeding. We noticed that, at taxonomic levels, the number of operational taxonomic units (OTUs), along with the Chao and Shannon indexes, significantly shifted in HFD-fed mice compared to those fed a ND. Similarly, at the phylum level, an increase in Firmicutes and a decrease in Bacteroidetes were noticed in HFD-fed mice. At the genus level, an increase in Lactobacillus and Ruminococcus was observed, while Allobaculum, Clostridium, and Akkermansia were markedly reduced in the HFD group. Many bacteria from the Ruminococcus genus impair bile acid metabolism and restrict weight loss. Firmicutes are efficient in breaking down complex carbohydrates into short-chain fatty acids (SCFAs) and other metabolites, whereas Bacteroidetes are involved in a more balanced or efficient energy extraction. Thus, an increase in Firmicutes over Bacteroidetes enhances the absorption of more calories from food, which may contribute to obesity. Taken together, the altered gut microbiota and metabolites trigger AT inflammation, which contributes to metabolic dysregulation and disease progression. Thus, this study highlights the potential of the gut microbiome in the development of therapeutic strategies for obesity and related metabolic disorders.
Keywords: dysbiosis; gut microbiota; metabolic profile; metabolites; obesity.
Conflict of interest statement
All the authors declare that they have no competing interests that could have influenced this study and have not been involved with any study sponsors.
Figures



References
-
- Turnbaugh P.J., Ley R.E., Mahowald M.A., Magrini V., Mardis E.R., Gordon J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. - PubMed
-
- Blüher M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019;15:288–298. - PubMed
-
- Gu Y., Liu C., Zheng N., Jia W., Zhang W., Li H. Metabolic and gut microbial characterization of obesity-prone mice under a high-fat diet. J. Proteome Res. 2019;18:1703–1714. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
