

Multimodal SARS-CoV-2 interactome sketches the virus-host spatial organization
- PMID: 40140549
- PMCID: PMC11947133
- DOI: 10.1038/s42003-025-07933-z
Multimodal SARS-CoV-2 interactome sketches the virus-host spatial organization
Abstract
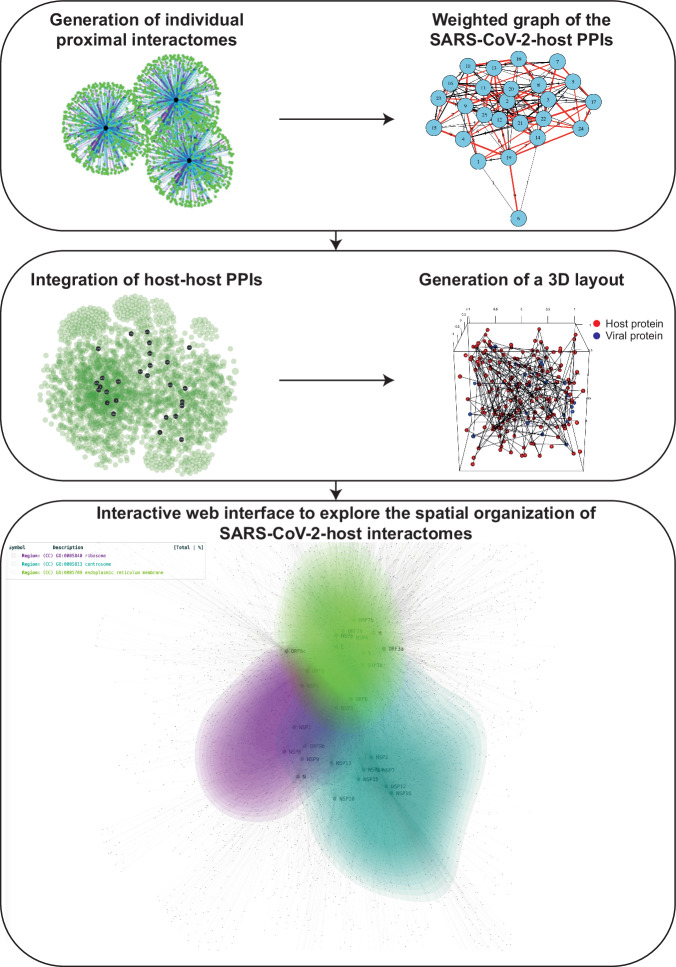
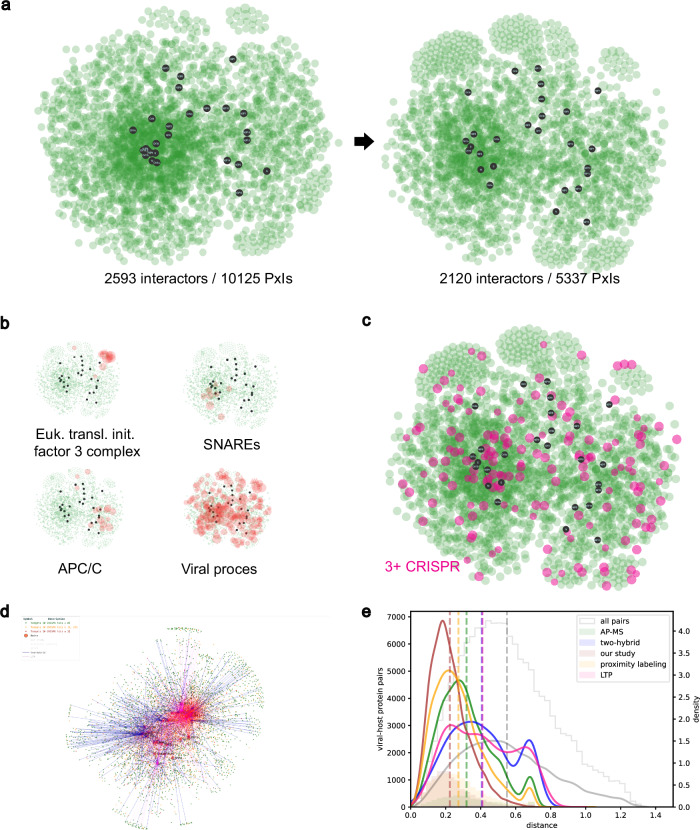
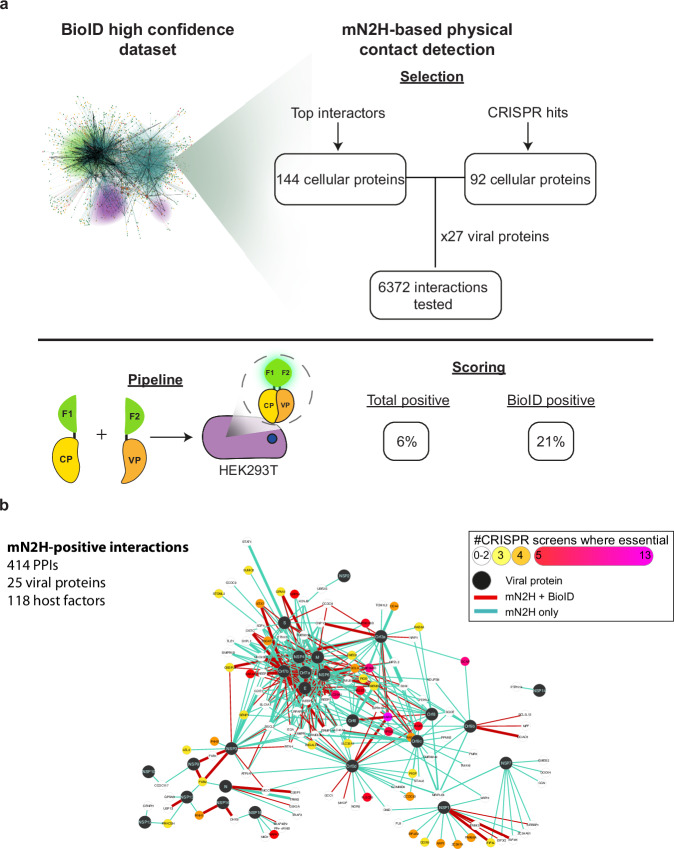
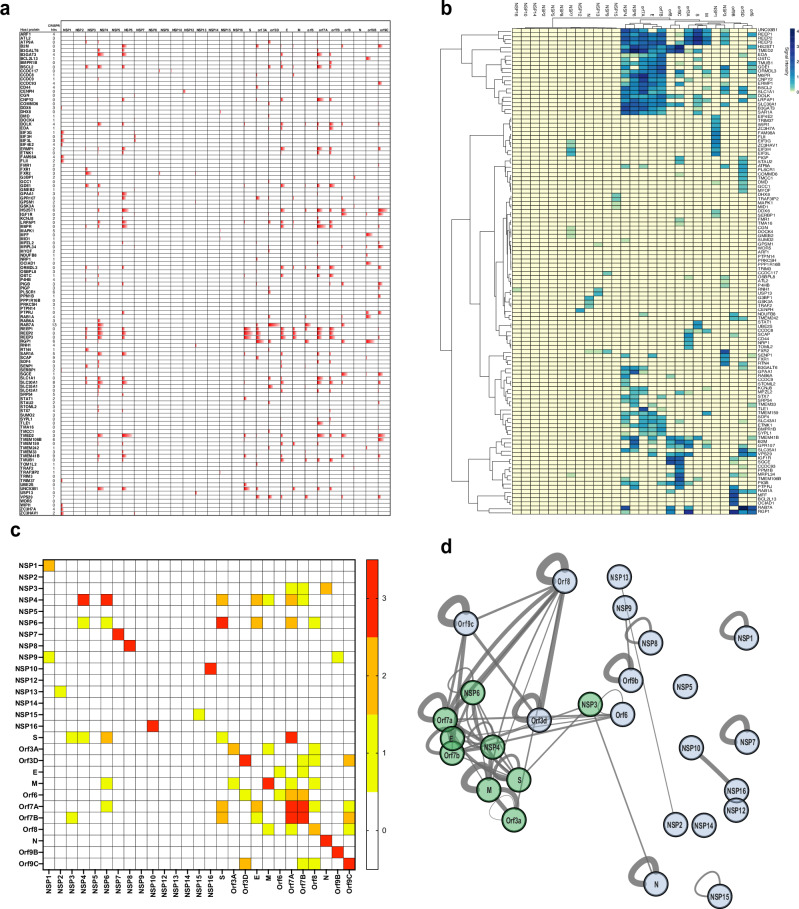
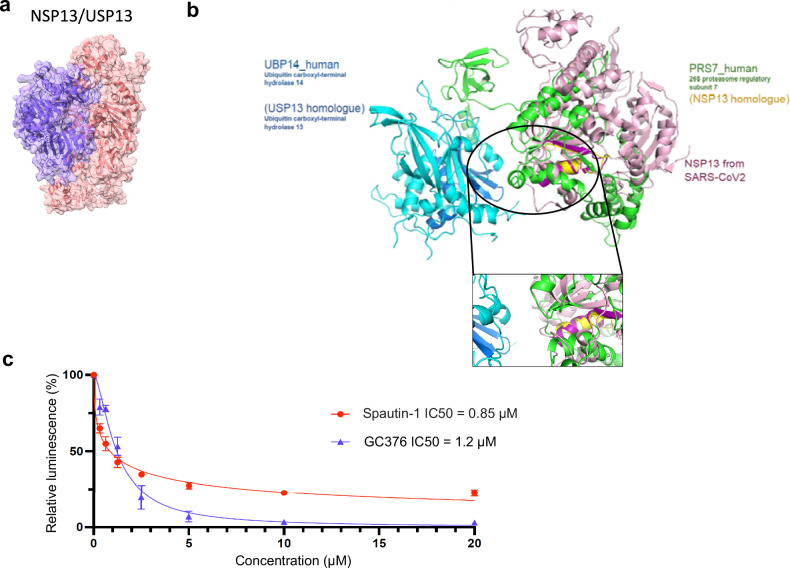
An accurate spatial representation of protein-protein interaction networks is needed to achieve a realistic and biologically relevant representation of interactomes. Here, we leveraged the spatial information included in Proximity-Dependent Biotin Identification (BioID) interactomes of SARS-CoV-2 proteins to calculate weighted distances and model the organization of the SARS-CoV-2-human interactome in three dimensions (3D) within a cell-like volume. Cell regions with viral occupancy were highlighted, along with the coordination of viral proteins exploiting the cellular machinery. Profiling physical intra-virus and virus-host contacts enabled us to demonstrate both the accuracy and the predictive value of our 3D map for direct interactions, meaning that proteins in closer proximity tend to interact physically. Several functionally important virus-host complexes were detected, and robust structural models were obtained, opening the way to structure-directed drug discovery screens. This PPI discovery pipeline approach brings us closer to a realistic spatial representation of interactomes, which, when applied to viruses or other pathogens, can provide significant information for infection. Thus, it represents a promising tool for coping with emerging infectious diseases.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures

References
-
- Drewes, G. & Bantscheff, M. Chemical Proteomics: Methods and Protocols (Humana Press, 2011).
-
- Link, A. J. et al. Targeted Identification of Protein Interactions in Eukaryotic mRNA Translation. Proteomics20, e1900177 (2020). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
