

Proteostasis and lysosomal repair deficits in transdifferentiated neurons of Alzheimer's disease
- PMID: 40140603
- PMCID: PMC11991917
- DOI: 10.1038/s41556-025-01623-y
Proteostasis and lysosomal repair deficits in transdifferentiated neurons of Alzheimer's disease
Abstract
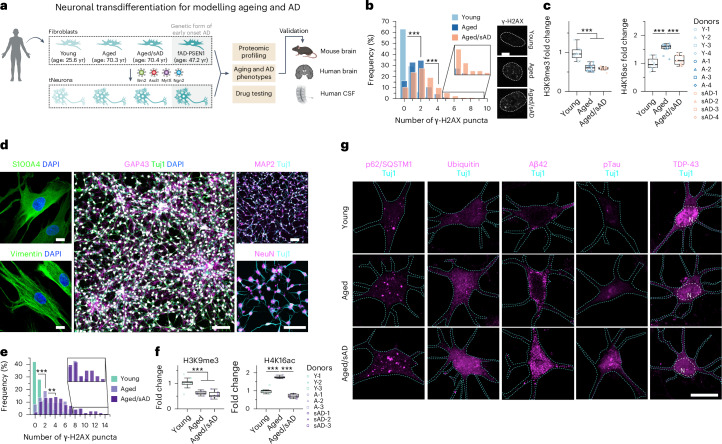
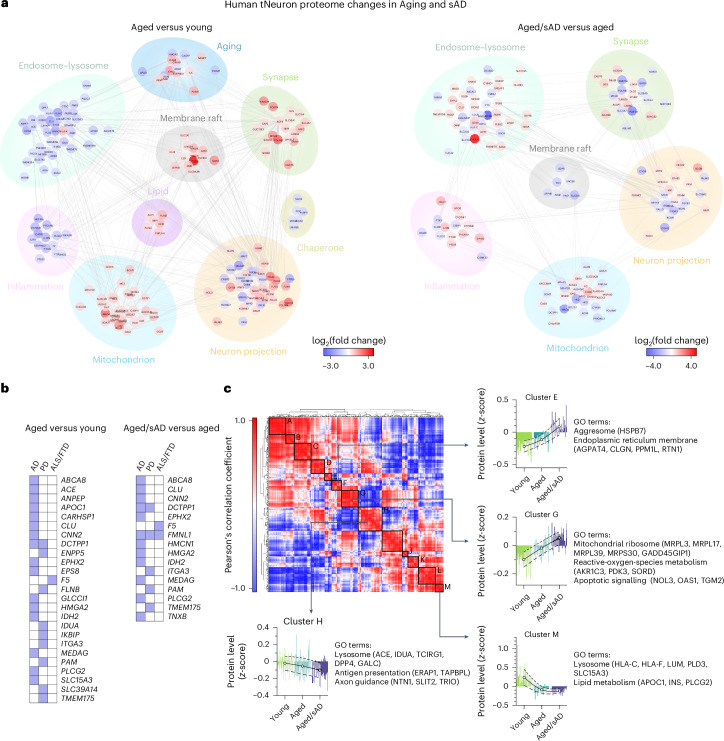
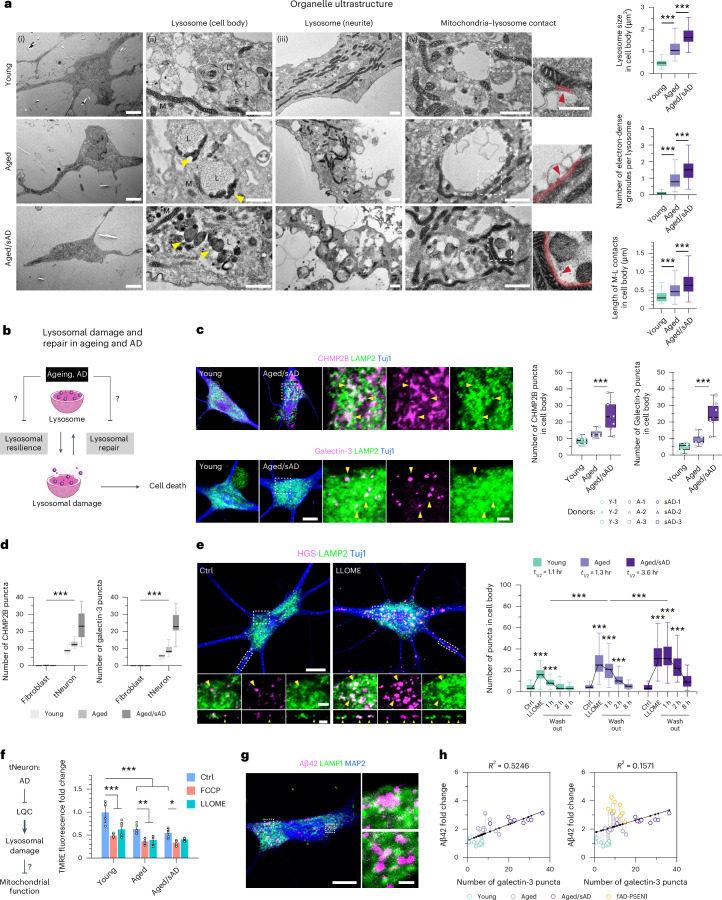
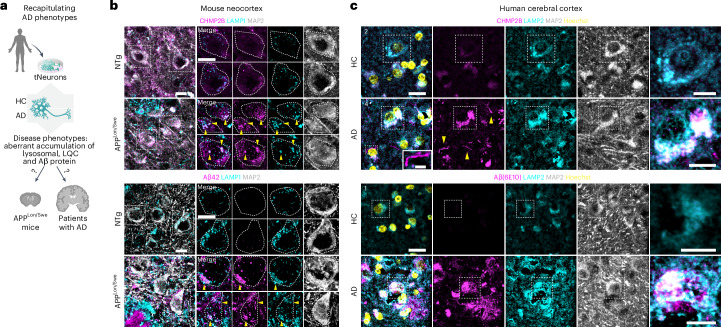
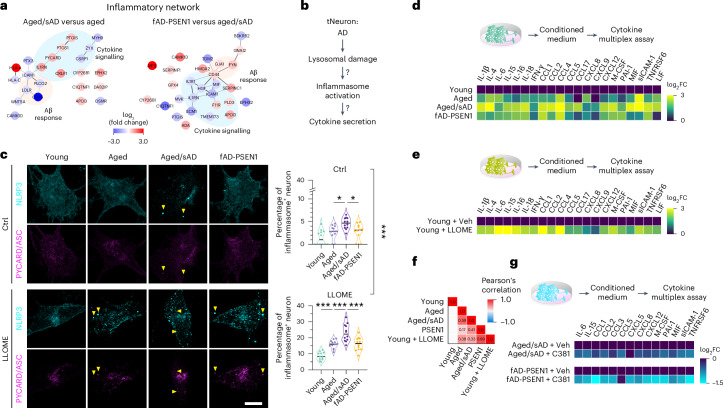
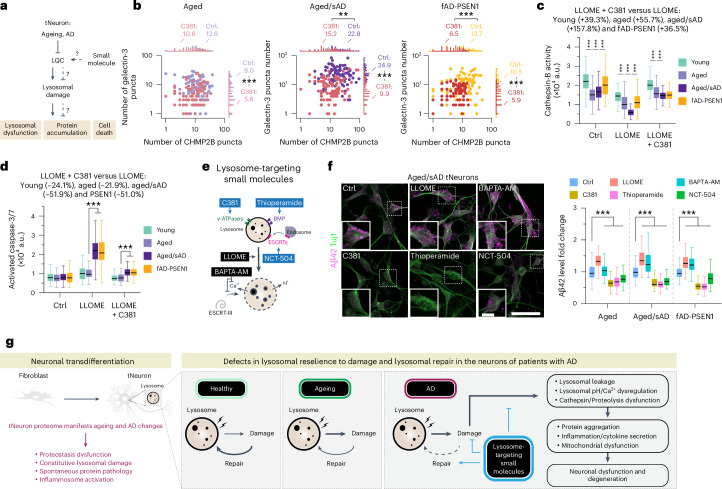
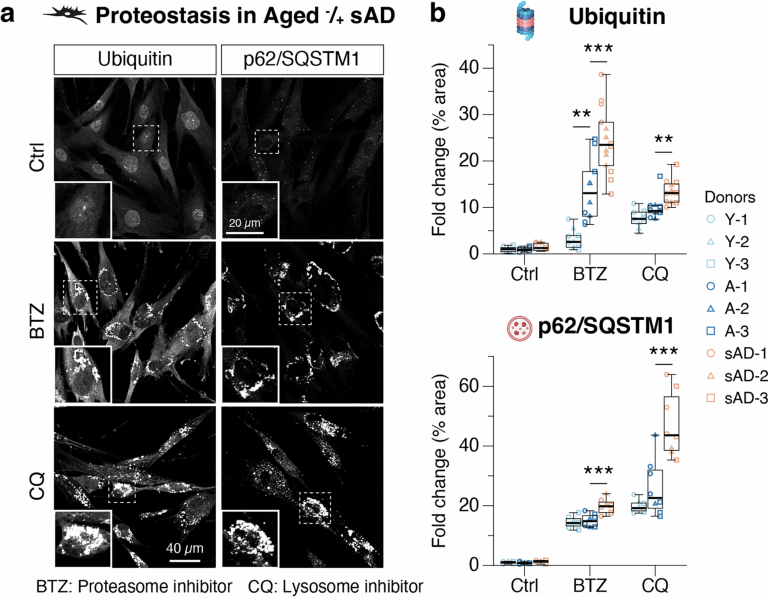
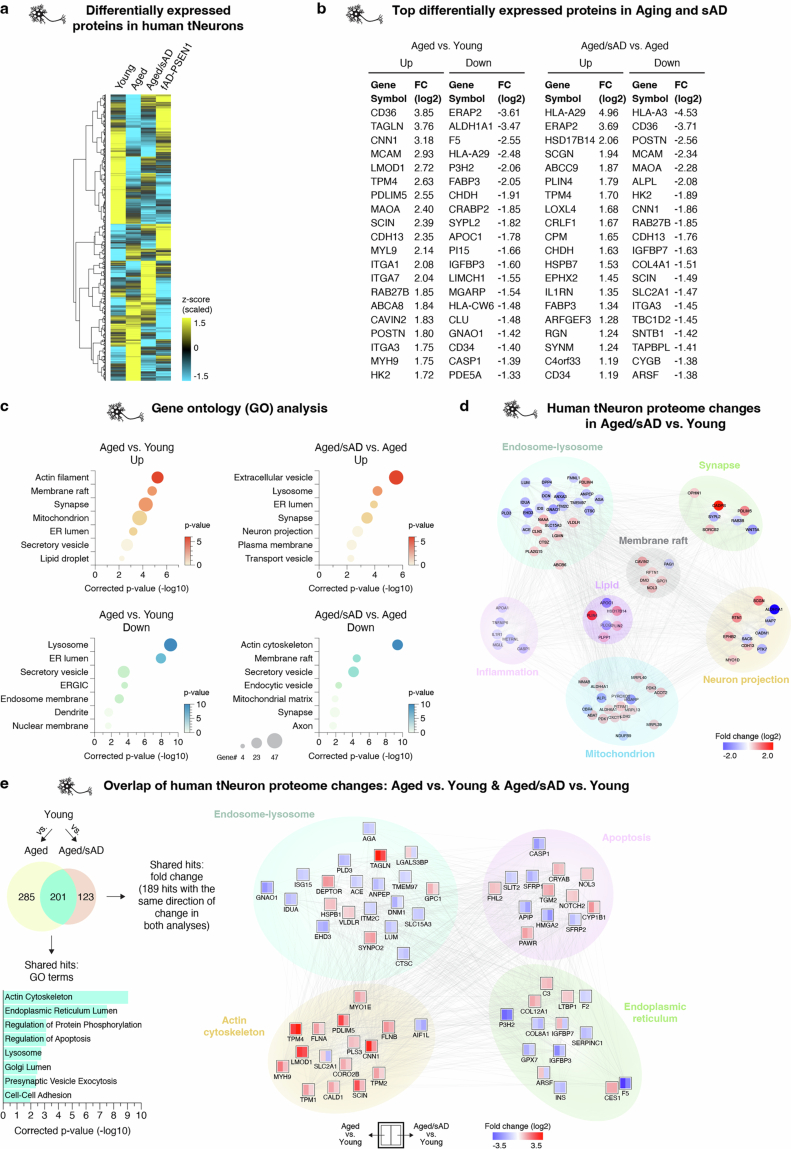
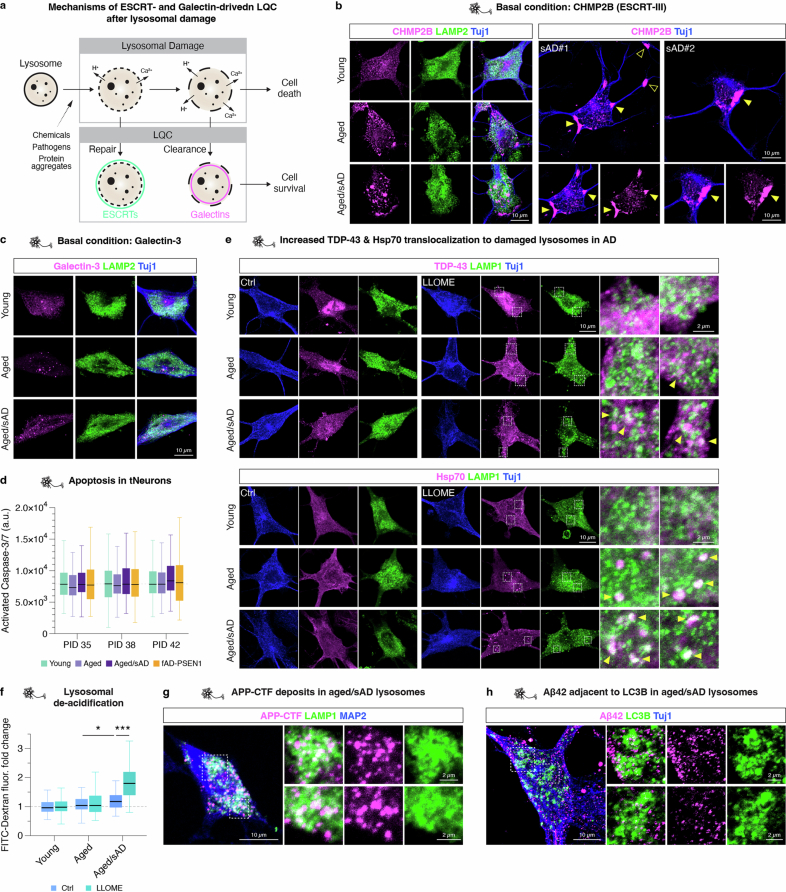
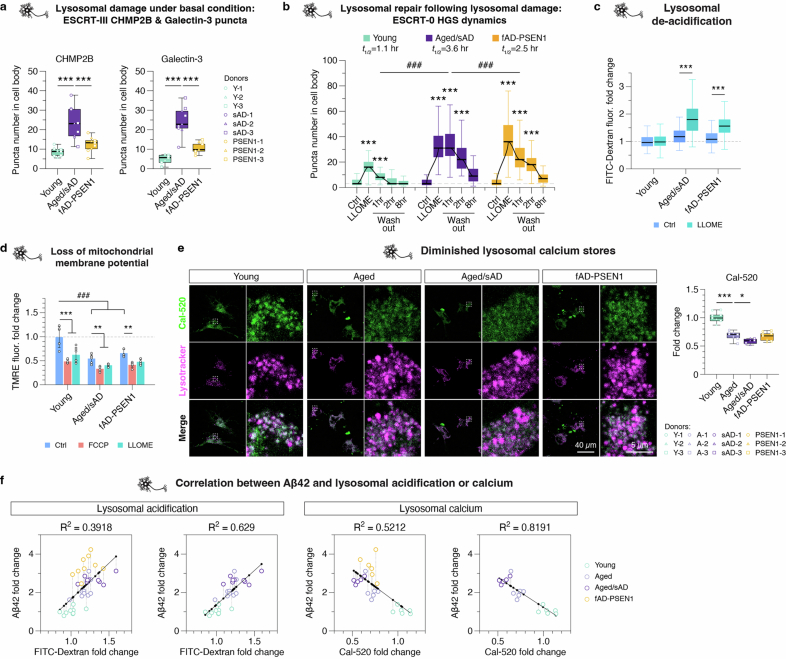
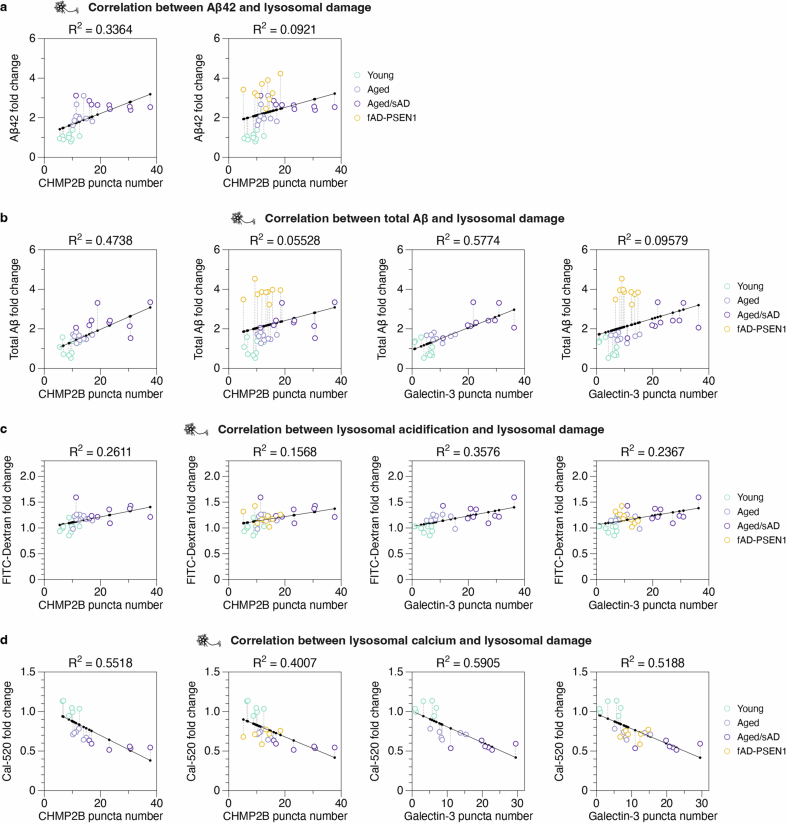
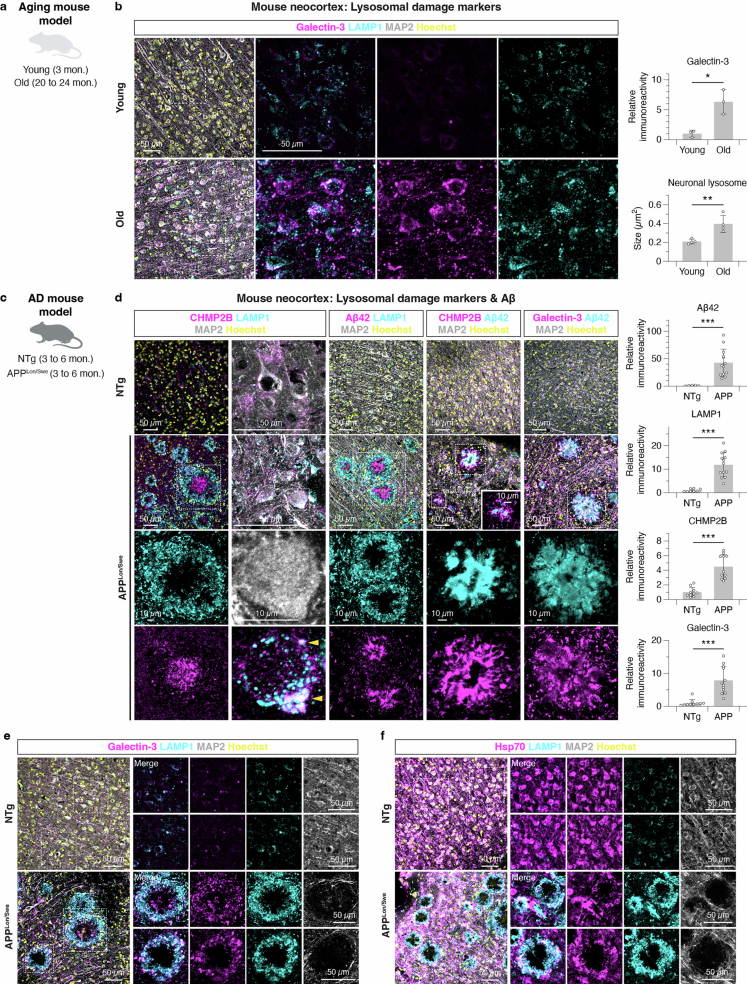
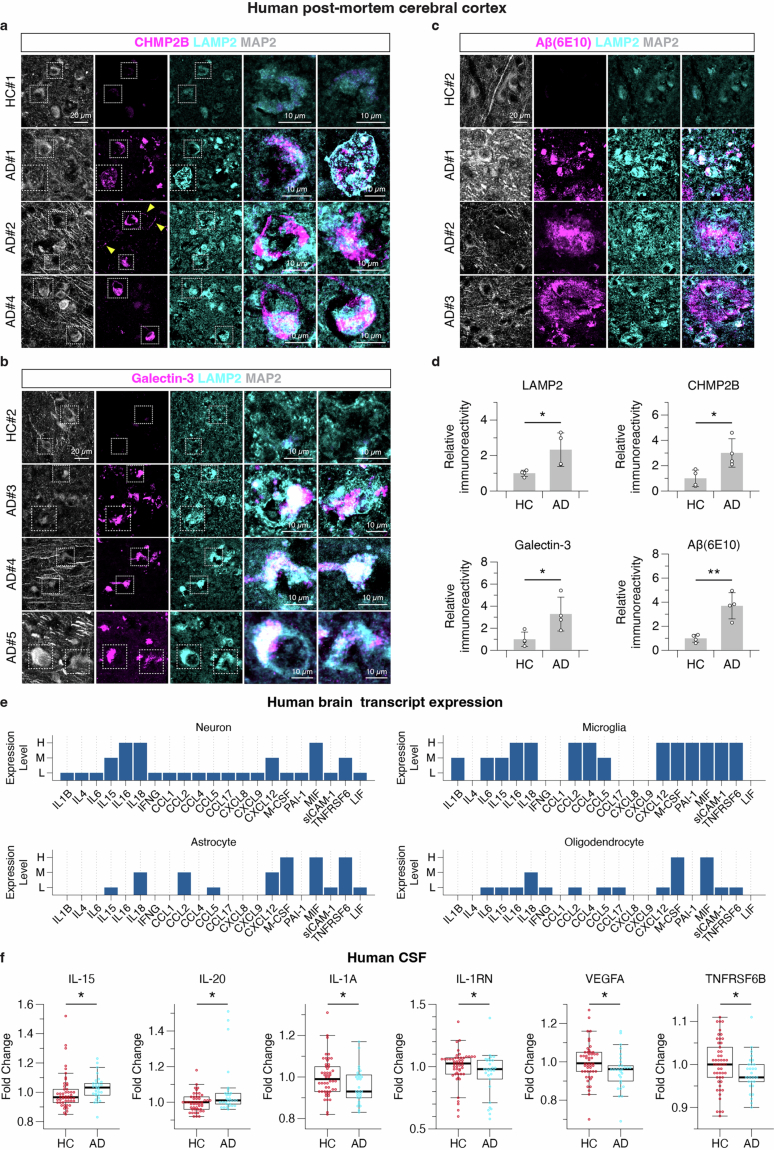
Ageing is the most prominent risk factor for Alzheimer's disease (AD). However, the cellular mechanisms linking neuronal proteostasis decline to the characteristic aberrant protein deposits in the brains of patients with AD remain elusive. Here we develop transdifferentiated neurons (tNeurons) from human dermal fibroblasts as a neuronal model that retains ageing hallmarks and exhibits AD-linked vulnerabilities. Remarkably, AD tNeurons accumulate proteotoxic deposits, including phospho-tau and amyloid β, resembling those in APP mouse brains and the brains of patients with AD. Quantitative tNeuron proteomics identify ageing- and AD-linked deficits in proteostasis and organelle homeostasis, most notably in endosome-lysosomal components. Lysosomal deficits in aged tNeurons, including constitutive lysosomal damage and ESCRT-mediated lysosomal repair defects, are exacerbated in AD tNeurons and linked to inflammatory cytokine secretion and cell death. Providing support for the centrality of lysosomal deficits in AD, compounds ameliorating lysosomal function reduce amyloid β deposits and cytokine secretion. Thus, the tNeuron model system reveals impaired lysosomal homeostasis as an early event of ageing and AD.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: R.V., J.L. and T.W.-C. are co-founders of Qinotto Inc. The other authors declare no competing interests.
Figures



Update of
-
Proteostasis and lysosomal repair deficits in transdifferentiated neurons of Alzheimer's disease.bioRxiv [Preprint]. 2025 Jan 13:2023.03.27.534444. doi: 10.1101/2023.03.27.534444. bioRxiv. 2025. Update in: Nat Cell Biol. 2025 Apr;27(4):619-632. doi: 10.1038/s41556-025-01623-y. PMID: 37034684 Free PMC article. Updated. Preprint.
-
Human tNeurons reveal aging-linked proteostasis deficits driving Alzheimer's phenotypes.Res Sq [Preprint]. 2024 May 30:rs.3.rs-4407236. doi: 10.21203/rs.3.rs-4407236/v1. Res Sq. 2024. Update in: Nat Cell Biol. 2025 Apr;27(4):619-632. doi: 10.1038/s41556-025-01623-y. PMID: 38853828 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
- P30 AG066515/AG/NIA NIH HHS/United States
- ASAP-000282/Michael J. Fox Foundation for Parkinson's Research (Michael J. Fox Foundation)
- P50 AG047366/AG/NIA NIH HHS/United States
- P01 AG054407/AG/NIA NIH HHS/United States
- AG054407/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)
LinkOut - more resources
Full Text Sources
Medical
Research Materials
