

Metabolic modelling reveals the aging-associated decline of host-microbiome metabolic interactions in mice
- PMID: 40140706
- PMCID: PMC11964932
- DOI: 10.1038/s41564-025-01959-z
Metabolic modelling reveals the aging-associated decline of host-microbiome metabolic interactions in mice
Abstract
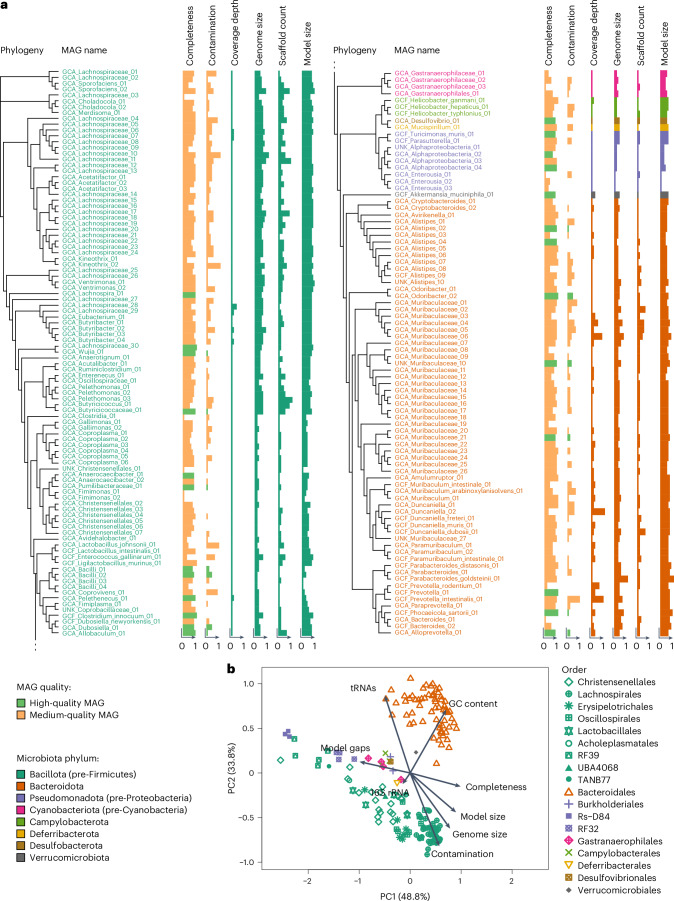
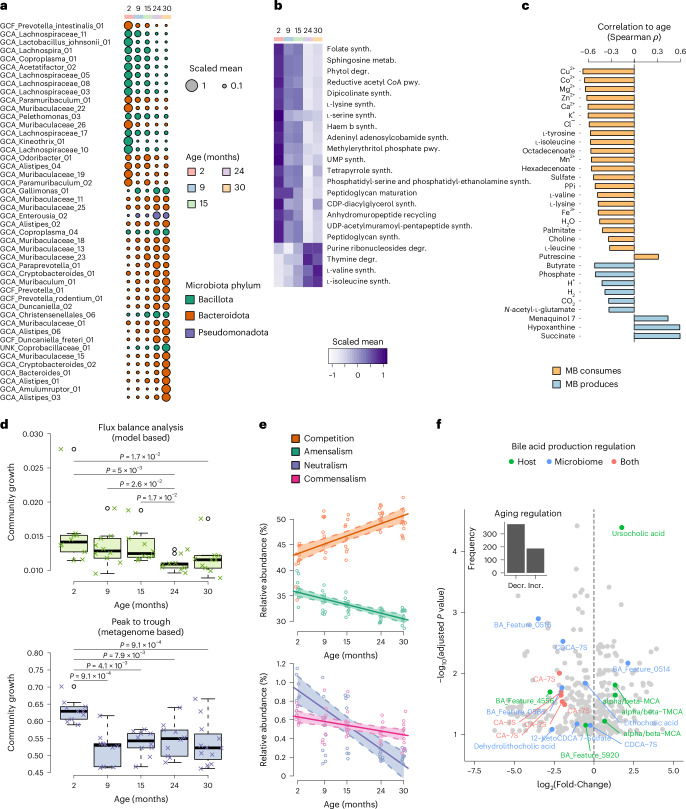
Aging is accompanied by considerable changes in the gut microbiome, yet the molecular mechanisms driving aging and the role of the microbiome remain unclear. Here we combined metagenomics, transcriptomics and metabolomics from aging mice with metabolic modelling to characterize host-microbiome interactions during aging. Reconstructing integrated metabolic models of host and 181 mouse gut microorganisms, we show a complex dependency of host metabolism on known and previously undescribed microbial interactions. We observed a pronounced reduction in metabolic activity within the aging microbiome accompanied by reduced beneficial interactions between bacterial species. These changes coincided with increased systemic inflammation and the downregulation of essential host pathways, particularly in nucleotide metabolism, predicted to rely on the microbiota and critical for preserving intestinal barrier function, cellular replication and homeostasis. Our results elucidate microbiome-host interactions that potentially influence host aging processes. These pathways could serve as future targets for the development of microbiome-based anti-aging therapies.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures











References
-
- Lopez, A. D., Mathers, C. D., Ezzati, M., Jamison, D. T. & Murray, C. J. L. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet367, 1747–1757 (2006). - PubMed
-
- López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: an expanding universe. Cell186, 243–278 (2023). - PubMed
-
- Zhou, Y., Hu, G. & Wang, M. C. Host and microbiota metabolic signals in aging and longevity. Nat. Chem. Biol.17, 1027–1036 (2021). - PubMed
MeSH terms
Grants and funding
- 859890/EC | Horizon 2020 Framework Programme (EU Framework Programme for Research and Innovation H2020)
- FOR5042/Deutsche Forschungsgemeinschaft (German Research Foundation)
- EXC2167/Deutsche Forschungsgemeinschaft (German Research Foundation)
- 416 418087534/Deutsche Forschungsgemeinschaft (German Research Foundation)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
