

Functional characterization of two KCND3 variants associated with SCA 19/22 ataxia in Latin American families
- PMID: 40140957
- PMCID: PMC11938557
- DOI: 10.1186/s40659-025-00589-3
Functional characterization of two KCND3 variants associated with SCA 19/22 ataxia in Latin American families
Abstract
Background: Spinocerebellar ataxia 19/22 (SCA19/22) represents a rare autosomal dominant genetic disorder resulting in progressive ataxia and cerebellar atrophy. SCA19/22 is caused by variants in the KCND3 gene, which encodes a voltage-gated potassium channel subunit essential for cerebellar Purkinje cell function. To date, 22 variants have been reported worldwide, with incomplete functional studies.
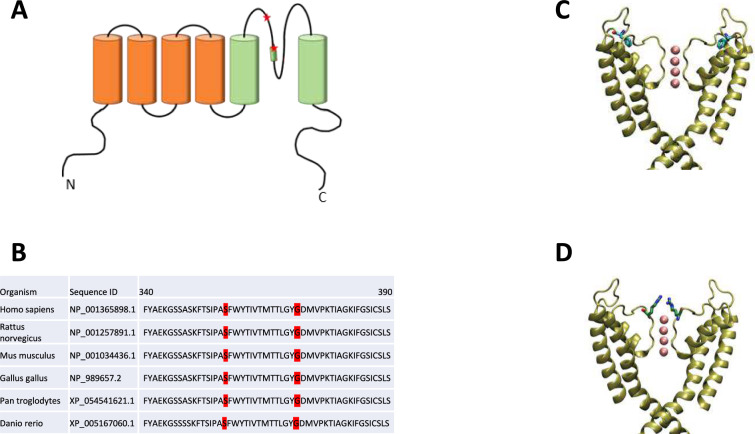
Results: We present four Chilean and Mexican cases in whom two single-nucleotide variants were identified through whole-exome sequencing of the probands. One variant (G371R) was initially cataloged as pathogenic and the other (S357W) as likely pathogenic according to the American College of Medical Genetics and Genomics criteria. The pathogenicity of the G371R variation was confirmed by in-silico mutagenesis. Our molecular models, that include electrostatic potential analysis and algorithms to analyze the pore dimensions (HOLE), indicated that the longer side chain of the arginine narrowed the channel's selectivity filter, while the positive charge modified its surface electrostatic potential, presumably preventing potassium flux. Functional characterization of the S357W variant was performed in AD293 cells. When overexpressed, KV4.3S357W channels alone showed no current. Protein electrophoresis revealed that the total number of KV4.3 channels expressed did not differ between the wild-type and mutated phenotypes, suggesting a protein trafficking malfunction. Co-expression of the KChIP2 auxiliary subunit partially rescued the potassium currents when the variant was expressed, albeit with very different biophysical characteristics, including faster inactivation vs. wild-type channels.
Conclusions: This functional characterization of two KCND3 variants associated with SCA19/22 adds new evidence for the pathogenic role of Kv4.3 loss-of-function mutations and establishes a correlation between functional dominance and clinical severity in SCA19/22.
Keywords: KCND3; Congenital ataxia; Functional characterization; Kv4.3; Spinocerebellar ataxia SCA19/22.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: This study was performed in line with the principles of the Declaration of Helsinki. All participants provided informed consent for the genetic study and the publication of their clinical information. Approval was granted by the Investigation in Human Beings Ethics Committee of Facultad de Medicina-Universidad de Chile and carried out in compliance with the current legislation in Chile on Scientific Research in humans of laws 20120, and 19,628. Consent for publication: All authors approved the final version of the manuscript. Competing interests: The authors declare that they have no competing interests.
Figures




References
-
- Hsiao CT, Fu SJ, Liu YT, Lu YH, Zhong CY, Tang CY, et al. Novel SCA19/22-associated KCND3 mutations disrupt human K(V) 4.3 protein biosynthesis and channel gating. Hum Mutat. 2019;40(11):2088–107. - PubMed
-
- Avila-Jaque D, Martin F, Bustamante ML, Luna Alvarez M, Fernandez JM, Ortiz D, de Montellano DJ, et al. The phenotypic spectrum of spinocerebellar ataxia type 19 in a series of Latin American patients. Cerebellum. 2024;23(4):1727–32. - PubMed
-
- Klockgether T, Mariotti C, Paulson HL. Spinocerebellar ataxia. Nat Rev Dis Primers. 2019;5(1):24. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
