

ONT in Clinical Diagnostics of Repeat Expansion Disorders: Detection and Reporting Challenges
- PMID: 40141365
- PMCID: PMC11942491
- DOI: 10.3390/ijms26062725
ONT in Clinical Diagnostics of Repeat Expansion Disorders: Detection and Reporting Challenges
Abstract
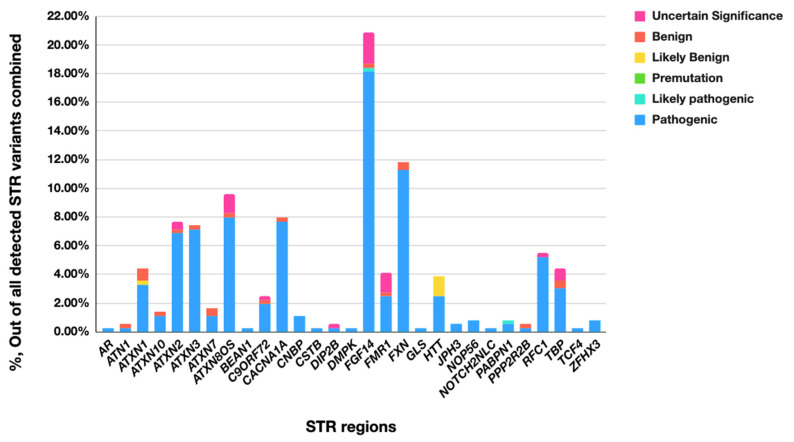
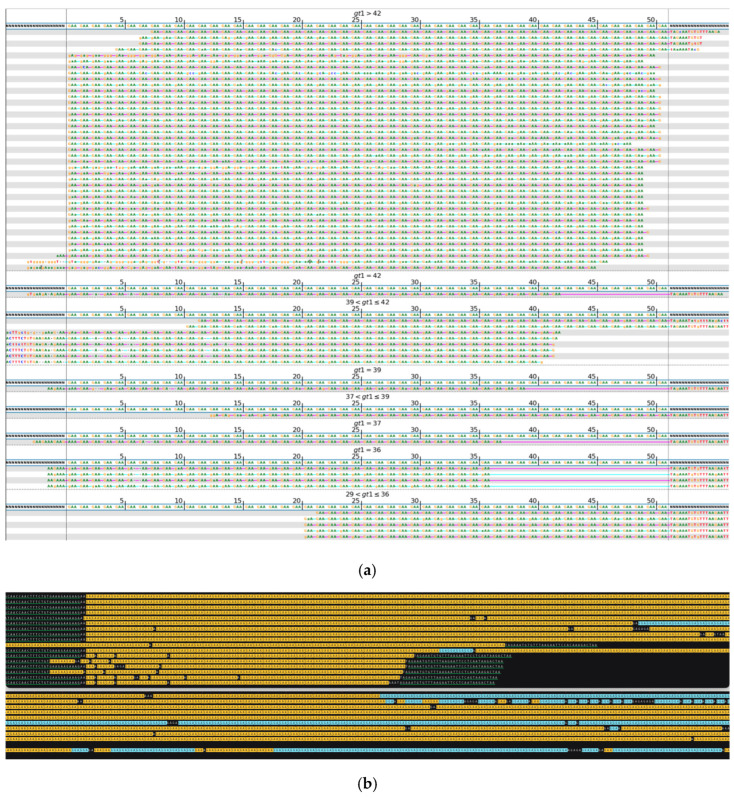
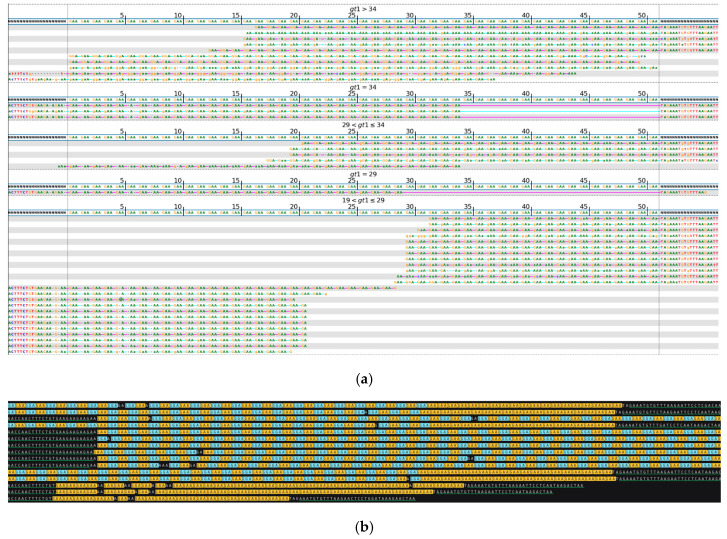
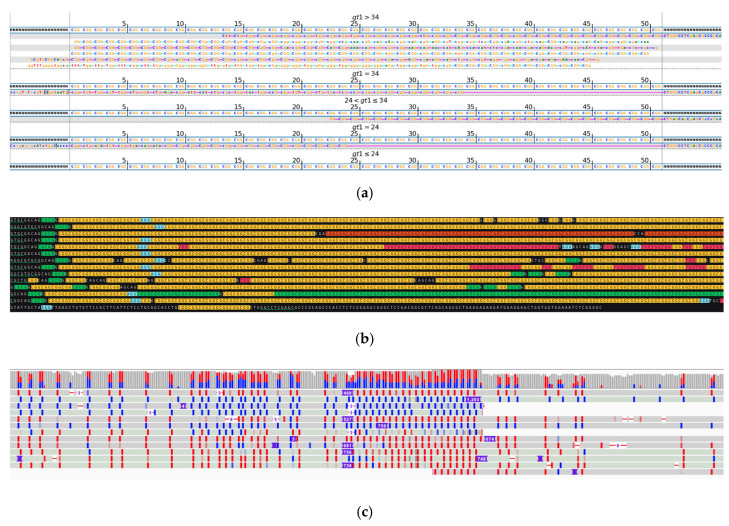
While whole-genome sequencing (WGS) using short-read technology has become a standard diagnostic test, this technology has limitations in analyzing certain genomic regions, particularly short tandem repeats (STRs). These repetitive sequences are associated with over 50 diseases, primarily affecting neurological function, including Huntington disease, frontotemporal dementia, and Friedreich's ataxia. We analyzed 2689 cases with movement disorders and dementia-related phenotypes processed at Variantyx in 2023-2024 using a two-tiered approach, with an initial short-read WGS followed by ONT long-read sequencing (when necessary) for variant characterization. Of the 2038 cases (75.8%) with clinically relevant genetic variants, 327 (16.0%) required additional long-read analysis. STR variants were reported in 338 cases (16.6% of positive cases), with approximately half requiring long-read sequencing for definitive classification. The combined approach enabled the precise determination of repeat length, composition, somatic mosaicism, and methylation status. Notable advantages included the detection of complex repeat structures in several genes such as RFC1, FGF14, and FXN, where long-read sequencing allowed to determine somatic repeat unit variations and accurate allele phasing. Further studies are needed to establish technology-specific guidelines for the standardized interpretation of long-read sequencing data for the clinical diagnostics of repeat expansion disorders.
Keywords: Nanopore; ONT; WGS; ataxia; dementia; genetic testing; long reads; repeat expansion.
Conflict of interest statement
Authors Ludmila Kaplun, Greice Krautz-Peterson, Nir Neerman, Yocheved Schindler, Elinor Dehan, Claudia S. Huettner, Brett K. Baumgartner, Christine Stanley, and Alexander Kaplun were employed by the company Variantyx, Framingham, MA 01701, USA.
Figures



Similar articles
-
An update on the neurological short tandem repeat expansion disorders and the emergence of long-read sequencing diagnostics.Acta Neuropathol Commun. 2021 May 25;9(1):98. doi: 10.1186/s40478-021-01201-x. Acta Neuropathol Commun. 2021. PMID: 34034831 Free PMC article. Review.
-
ONT long-read WGS for variant discovery and orthogonal confirmation of short read WGS derived genetic variants in clinical genetic testing.Front Genet. 2023 Apr 21;14:1145285. doi: 10.3389/fgene.2023.1145285. eCollection 2023. Front Genet. 2023. PMID: 37152986 Free PMC article.
-
Long-read sequencing across the C9orf72 'GGGGCC' repeat expansion: implications for clinical use and genetic discovery efforts in human disease.Mol Neurodegener. 2018 Aug 21;13(1):46. doi: 10.1186/s13024-018-0274-4. Mol Neurodegener. 2018. PMID: 30126445 Free PMC article.
-
Nanopore Long-Read Sequencing as a First-Tier Diagnostic Test to Detect Repeat Expansions in Neurological Disorders.Int J Mol Sci. 2025 Mar 21;26(7):2850. doi: 10.3390/ijms26072850. Int J Mol Sci. 2025. PMID: 40243408 Free PMC article.
-
Identification and characterization of repeat expansions in neurological disorders: Methodologies, tools, and strategies.Rev Neurol (Paris). 2024 May;180(5):383-392. doi: 10.1016/j.neurol.2024.03.005. Epub 2024 Apr 8. Rev Neurol (Paris). 2024. PMID: 38594146 Review.
Cited by
-
Long-Read Sequencing and Structural Variant Detection: Unlocking the Hidden Genome in Rare Genetic Disorders.Diagnostics (Basel). 2025 Jul 17;15(14):1803. doi: 10.3390/diagnostics15141803. Diagnostics (Basel). 2025. PMID: 40722552 Free PMC article. Review.
References
-
- Wigby K.M., Brockman D., Costain G., Hale C., Taylor S.L., Belmont J., Bick D., Dimmock D., Fernbach S., Greally J., et al. Evidence Review and Considerations for Use of First Line Genome Sequencing to Diagnose Rare Genetic Disorders. NPJ Genom. Med. 2024;9:15. doi: 10.1038/s41525-024-00396-x. - DOI - PMC - PubMed
-
- van der Sanden B.P.G.H., Schobers G., Corominas Galbany J., Koolen D.A., Sinnema M., van Reeuwijk J., Stumpel C.T.R.M., Kleefstra T., de Vries B.B.A., Ruiterkamp-Versteeg M., et al. The Performance of Genome Sequencing as a First-Tier Test for Neurodevelopmental Disorders. Eur. J. Human. Genet. 2022;31:81–88. doi: 10.1038/s41431-022-01185-9. - DOI - PMC - PubMed
-
- Rajan-Babu I.-S., Peng J.J., Chiu R., Birch P., Couse M., Guimond C., Lehman A., Mwenifumbo J., van Karnebeek C., Friedman J., et al. Genome-Wide Sequencing as a First-Tier Screening Test for Short Tandem Repeat Expansions. Genome Med. 2021;13:126. doi: 10.1186/s13073-021-00932-9. - DOI - PMC - PubMed
-
- Billingsley K.J., Meredith M., Daida K., Alvarez Jerez P., Negi S., Malik L., Genner R.M., Moller A., Zheng X., Gibson S.B., et al. Long-Read Sequencing of Hundreds of Diverse Brains Provides Insight into the Impact of Structural Variation on Gene Expression and DNA Methylation. Preprint. bioRxiv. 2024 doi: 10.1101/2024.12.16.628723. - DOI
-
- Gustafson J.A., Gibson S.B., Damaraju N., Zalusky M.P., Hoekzema K., Twesigomwe D., Yang L., Snead A.A., Richmond P.A., De Coster W., et al. High-Coverage Nanopore Sequencing of Samples from the 1000 Genomes Project to Build a Comprehensive Catalog of Human Genetic Variation. Genome Res. 2024;34:2061. doi: 10.1101/gr.279273.124. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
