

CDK-mediated phosphorylation of PNKP is required for end-processing of single-strand DNA gaps on Okazaki fragments and genome stability
- PMID: 40146629
- PMCID: PMC11949490
- DOI: 10.7554/eLife.99217
CDK-mediated phosphorylation of PNKP is required for end-processing of single-strand DNA gaps on Okazaki fragments and genome stability
Abstract
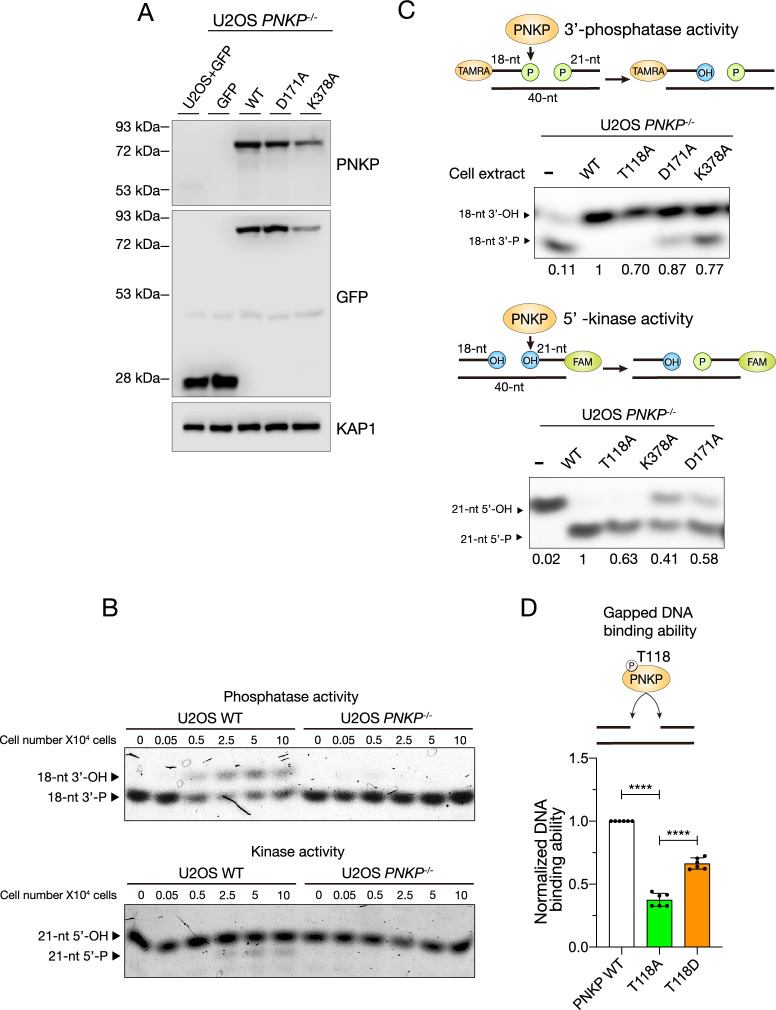
Polynucleotide kinase phosphatase (PNKP) has enzymatic activities as 3'-phosphatase and 5'-kinase of DNA ends to promote DNA ligation and repair. Here, we show that cyclin-dependent kinases (CDKs) regulate the phosphorylation of threonine 118 (T118) in PNKP. This phosphorylation allows recruitment to the gapped DNA structure found in single-strand DNA (ssDNA) nicks and/or gaps between Okazaki fragments (OFs) during DNA replication. T118A (alanine)-substituted PNKP-expressing cells exhibited an accumulation of ssDNA gaps in S phase and accelerated replication fork progression. Furthermore, PNKP is involved in poly (ADP-ribose) polymerase 1 (PARP1)-dependent replication gap filling as part of a backup pathway in the absence of OFs ligation. Altogether, our data suggest that CDK-mediated PNKP phosphorylation at T118 is important for its recruitment to ssDNA gaps to proceed with OFs ligation and its backup repairs via the gap-filling pathway to maintain genome stability.
Keywords: DNA repair; DNA replication; Okazaki fragment; PNKP; cell biology; human.
© 2025, Tsukada, Imamura et al.
Conflict of interest statement
KT, RI, TM, KS, MS, NK, LF, MI, YM, MS No competing interests declared
Figures














Update of
- doi: 10.1101/2021.07.29.452278
References
-
- Burhans WC, Vassilev LT, Wu J, Sogo JM, Nallaseth FS, DePamphilis ML. Emetine allows identification of origins of mammalian DNA replication by imbalanced DNA synthesis, not through conservative nucleosome segregation. The EMBO Journal. 1991;10:4351–4360. doi: 10.1002/j.1460-2075.1991.tb05013.x. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
