

Fabry Disease: Insights into Pathophysiology and Novel Therapeutic Strategies
- PMID: 40149601
- PMCID: PMC11940501
- DOI: 10.3390/biomedicines13030624
Fabry Disease: Insights into Pathophysiology and Novel Therapeutic Strategies
Abstract
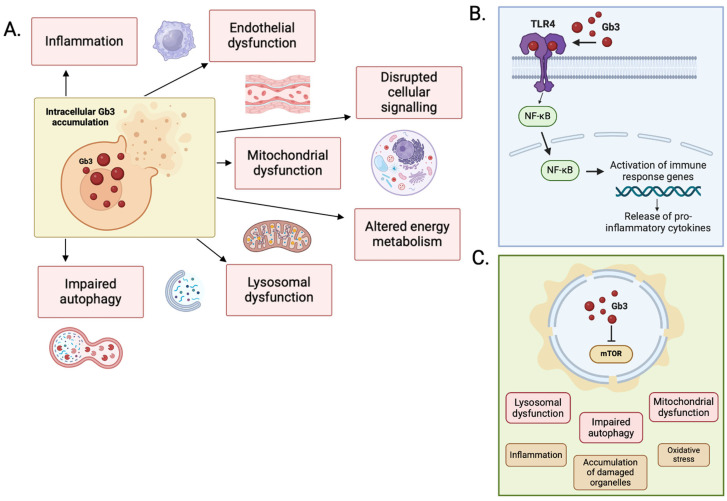
Fabry disease (FD) is an X-linked lysosomal storage disorder characterized by deficiency of α-galactosidase A (α-GalA), leading to the accumulation of glycosphingolipids and multi-organ dysfunction, particularly affecting the cardiovascular and renal systems. Disease-modifying treatments such as enzyme replacement therapy (ERT) and oral chaperone therapy (OCT) have limited efficacy, particularly in advanced disease, prompting a need for innovative therapeutic approaches targeting underlying molecular mechanisms beyond glycosphingolipid storage alone. Recent insights into the pathophysiology of FD highlights chronic inflammation and mitochondrial, lysosomal, and endothelial dysfunction as key mediators of disease progression. Adjunctive therapies such as sodium-glucose cotransporter-2 (SGLT2) inhibitors, glucagon-like peptide-1 (GLP-1) agonists, and mineralocorticoid receptor antagonists (MRAs) demonstrate significant cardiovascular and renal benefits in conditions including heart failure and chronic kidney disease. These drugs also modulate pathways involved in the pathophysiology of FD, such as autophagy, oxidative stress, and pro-inflammatory cytokine signaling. While theoretical foundations support their utility, dedicated trials are necessary to confirm efficacy in the FD-specific population. This narrative review highlights the importance of expanding therapeutic strategies in FD, advocating for a multi-faceted approach involving evidence-based adjunctive treatments to improve outcomes. Tailored research focusing on diverse FD phenotypes, including females and non-classical variants of disease, will be critical to advancing care and improving outcomes in this complex disorder.
Keywords: Fabry disease; chronic kidney disease; heart failure; inflammation.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures



Similar articles
-
Fabry Disease: Molecular Basis, Pathophysiology, Diagnostics and Potential Therapeutic Directions.Biomolecules. 2021 Feb 12;11(2):271. doi: 10.3390/biom11020271. Biomolecules. 2021. PMID: 33673160 Free PMC article. Review.
-
Anderson-Fabry disease: a multiorgan disease.Curr Pharm Des. 2013;19(33):5974-96. doi: 10.2174/13816128113199990352. Curr Pharm Des. 2013. PMID: 23448451 Review.
-
Combination therapy: an upcoming paradigm to improve kidney and cardiovascular outcomes in chronic kidney disease.Nephrol Dial Transplant. 2025 Feb 5;40(Supplement_1):i3-i17. doi: 10.1093/ndt/gfae212. Nephrol Dial Transplant. 2025. PMID: 39907543 Free PMC article. Review.
-
Could nutritional therapy take us further in our approaches to Fabry disease?Nutrition. 2020 Apr;72:110664. doi: 10.1016/j.nut.2019.110664. Epub 2019 Nov 29. Nutrition. 2020. PMID: 31972420 Review.
-
Complement activation and cellular inflammation in Fabry disease patients despite enzyme replacement therapy.Front Immunol. 2024 Jan 18;15:1307558. doi: 10.3389/fimmu.2024.1307558. eCollection 2024. Front Immunol. 2024. PMID: 38304433 Free PMC article.
Cited by
-
Real-world clinical outcomes in adult patients with Fabry disease: A 20-year retrospective observational cohort study from a single centre.Mol Genet Metab Rep. 2025 May 14;43:101229. doi: 10.1016/j.ymgmr.2025.101229. eCollection 2025 Jun. Mol Genet Metab Rep. 2025. PMID: 40485669 Free PMC article.
-
Fabry Disease Beyond Storage: The Role of Inflammation in Disease Progression.Int J Mol Sci. 2025 Jul 22;26(15):7054. doi: 10.3390/ijms26157054. Int J Mol Sci. 2025. PMID: 40806187 Free PMC article. Review.
References
-
- Biegstraaten M., Arngrímsson R., Barbey F., Boks L., Cecchi F., Deegan P.B., Feldt-Rasmussen U., Geberhiwot T., Germain D.P., Hendriksz C., et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: The European Fabry Working Group consensus document. Orphanet J. Rare Dis. 2015;10:36. doi: 10.1186/s13023-015-0253-6. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
