

Redox regulation of lung endothelial PERK, unfolded protein response (UPR) and proliferation via NOX1: Targeted inhibition as a potential therapy for PAH
- PMID: 40154102
- PMCID: PMC11986987
- DOI: 10.1016/j.redox.2025.103554
Redox regulation of lung endothelial PERK, unfolded protein response (UPR) and proliferation via NOX1: Targeted inhibition as a potential therapy for PAH
Abstract
Aims: Reactive oxygen species (ROS) play an important role in the pathogenesis of pulmonary arterial hypertension (PAH) and NADPH oxidases (NOXs) as sources of ROS are implicated in the development of the disease. We previously showed that NOX isozyme 1 (NOX1)-derived ROS contributes to pulmonary vascular endothelial cell (EC) proliferation in response to PAH triggers in vitro. However, whether and how NOX1 is involved in PAH in vivo have not been explored nor has NOX1 been examined as a viable and effective therapeutic disease target.
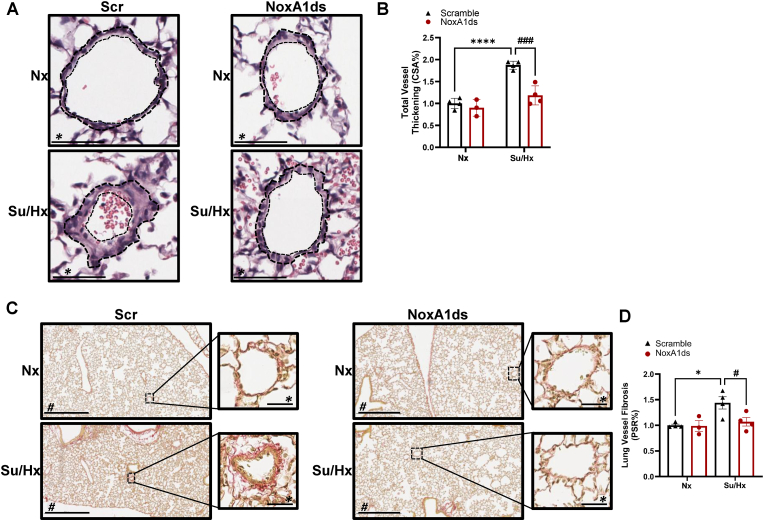
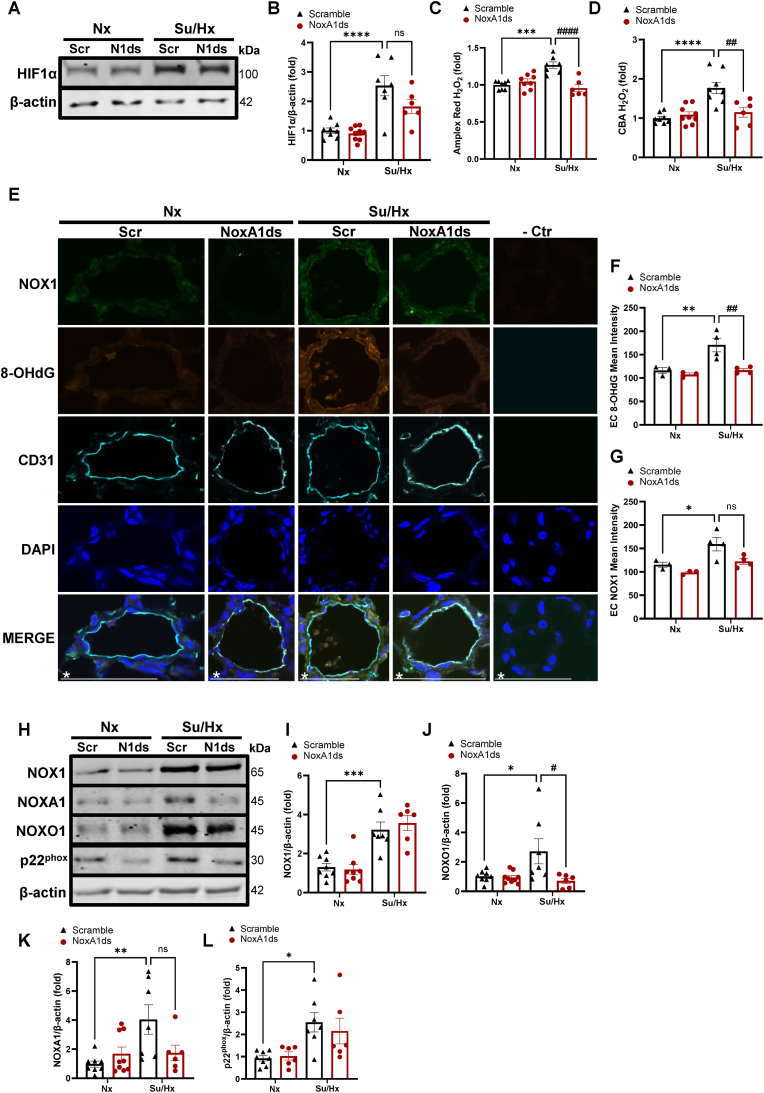
Methods and results: Herein, infusion of mice exposed to Sugen/hypoxia (10 % O2) with a specific NOX1 inhibitor, NOXA1ds, delivered via osmotic minipumps (i.p.), significantly suppressed pathological changes in hemodynamic parameters characteristic of PAH. Furthermore, lungs of human patients with idiopathic PAH (iPAH) and exploratory RNA-seq analysis of hypoxic human pulmonary ECs, in which NOX1 was suppressed, were probed. The findings showed a clear indication of NOX1 in the promotion of both protein disulfide isomerase (PDI) and the unfolded protein response (UPR; in particular, the PERK arm of the pathway including eIF2α and ATF4) leading to proliferation. In aggregate, these results are consistent with a causal role for NOX1 in the development of mouse and human PAH and reveal a novel and mechanistic pathway by which NOX1 activates the UPR response during EC proliferation.
Conclusion: NOX1 promotes phenotypic changes in ECs that are pivotal to proliferation and PAH through activation of the UPR. Taken together, our results are consistent with selective inhibition of NOX1 as a novel modality for attenuating PAH.
Keywords: Hypoxia; NADPH oxidases; NOX inhibitors; NOXA1ds; Pulmonary arterial hypertension; Unfolded protein response.
Copyright © 2025 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures






References
-
- Rawat D.K., Alzoubi A., Gupte R., Chettimada S., Watanabe M., Kahn A.G., Okada T., McMurtry I.F., Gupte S.A. Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure. Hypertension. 2014;64:1266–1274. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
