

A complex phylogeny of lineage plasticity in metastatic castration resistant prostate cancer
- PMID: 40155466
- PMCID: PMC11953479
- DOI: 10.1038/s41698-025-00854-4
A complex phylogeny of lineage plasticity in metastatic castration resistant prostate cancer
Abstract
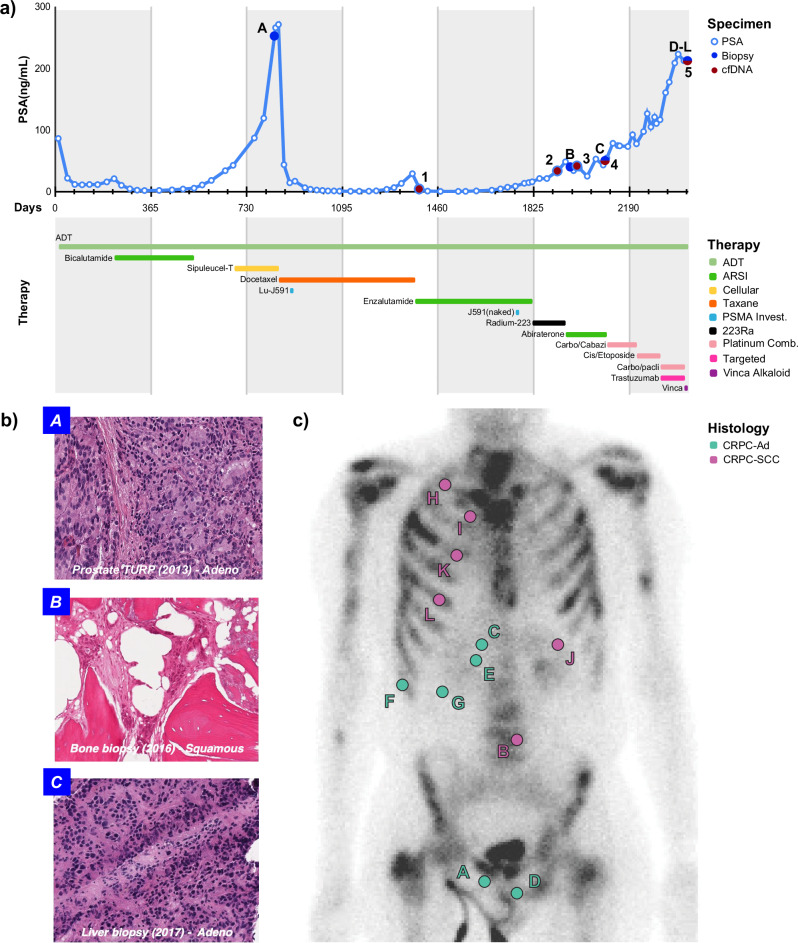
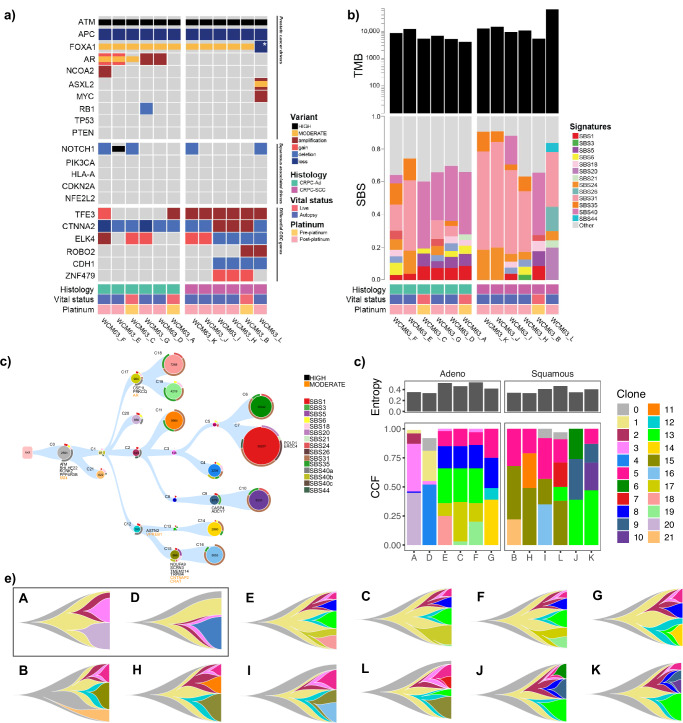
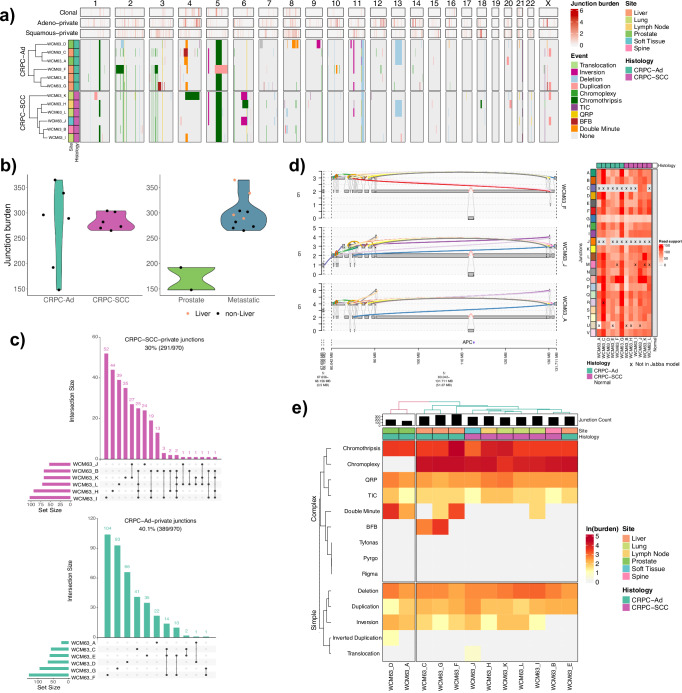
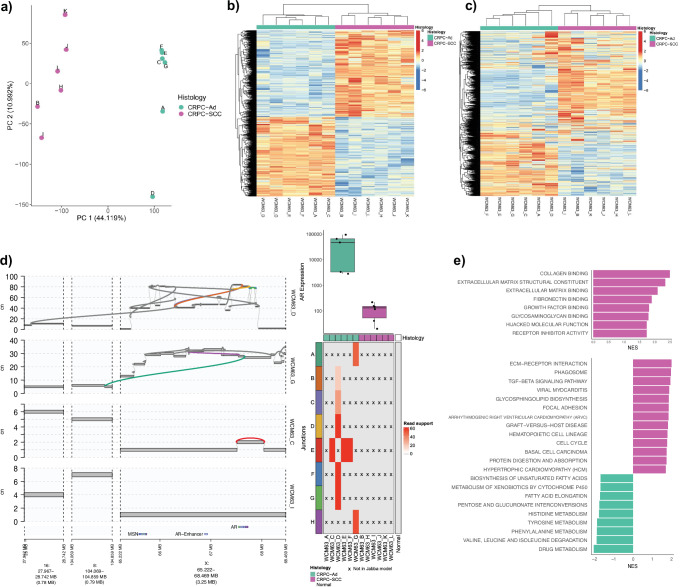
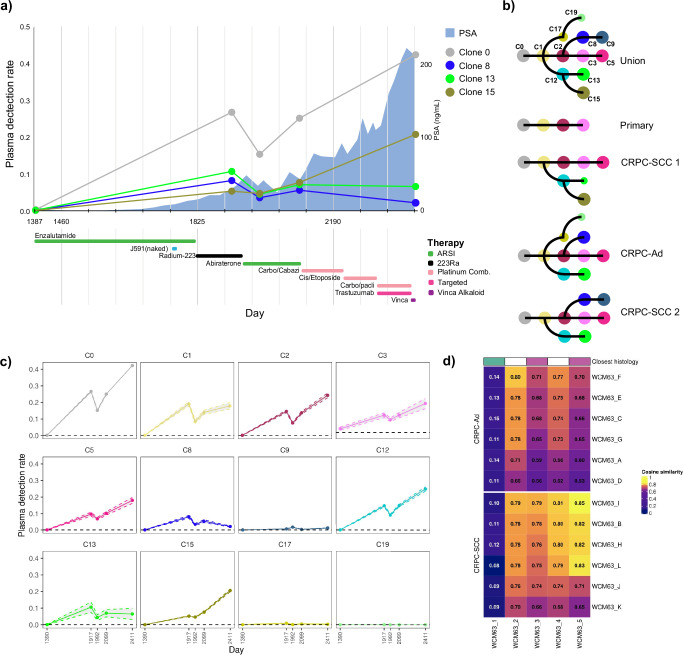
Aggressive variant and androgen receptor (AR)-independent castration resistant prostate cancers (CRPC) represent the most significant diagnostic and therapeutic challenges in prostate cancer. This study examined a case of simultaneous progression of both adenocarcinoma and squamous tumors from the same common origin. Using whole-genome and transcriptome sequencing from 17 samples collected over >6 years, we established the clonal relationship of all samples, defined shared complex structural variants, and demonstrated both divergent and convergent evolution at AR. Squamous CRPC-associated circulating tumor DNA was identified at clinical progression prior to biopsy detection of any squamous differentiation. Dynamic changes in the detection rate of histology-specific clones in circulation reflected histology-specific sensitivity to treatment. This dataset serves as an illustration of non-neuroendocrine transdifferentiation and highlights the importance of serial sampling at progression in CRPC for the detection of emergent non-adenocarcinoma histologies with implications for the treatment of lineage plasticity and transdifferentiation in metastatic CRPC.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: JTN reports consulting or advising relationships with Pfizer, Bayer, and AIQ. At the time of publication, he is an employee of Convergent Therapeutics.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
