

Cigarette smoke induces angiogenic activation in the cancer field through dysregulation of an endothelial microRNA
- PMID: 40155749
- PMCID: PMC11953391
- DOI: 10.1038/s42003-025-07710-y
Cigarette smoke induces angiogenic activation in the cancer field through dysregulation of an endothelial microRNA
Abstract
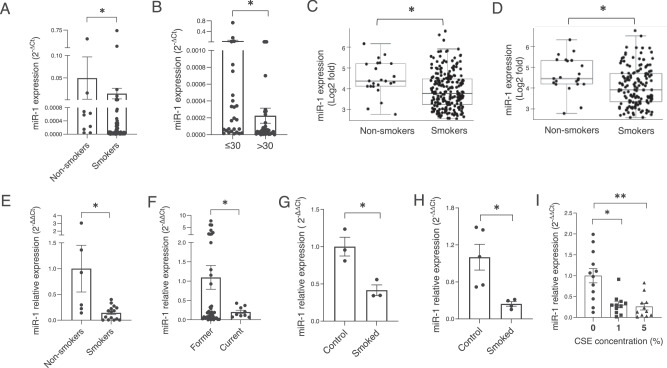
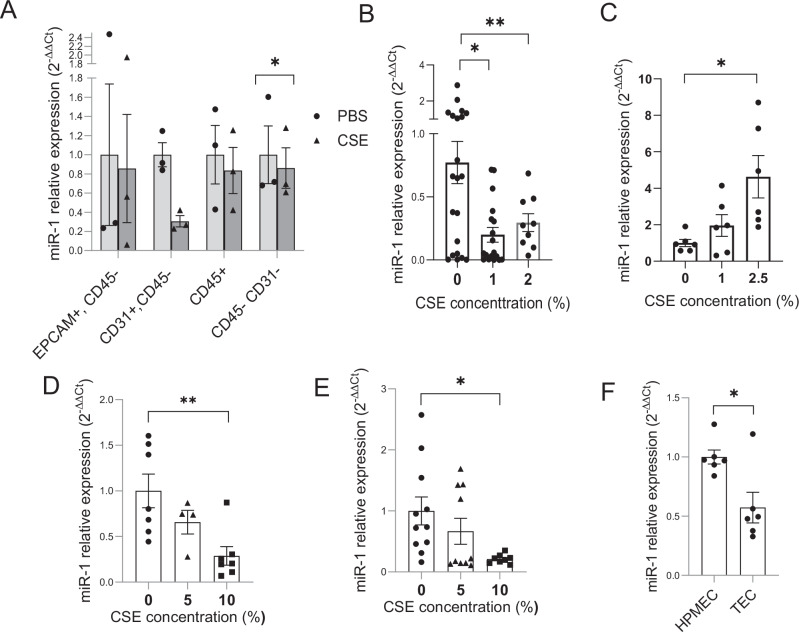
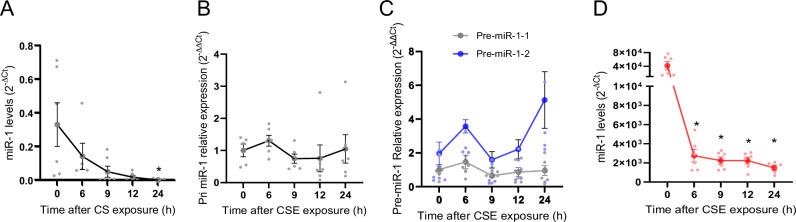
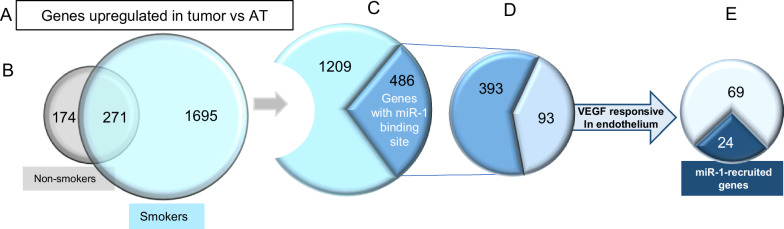
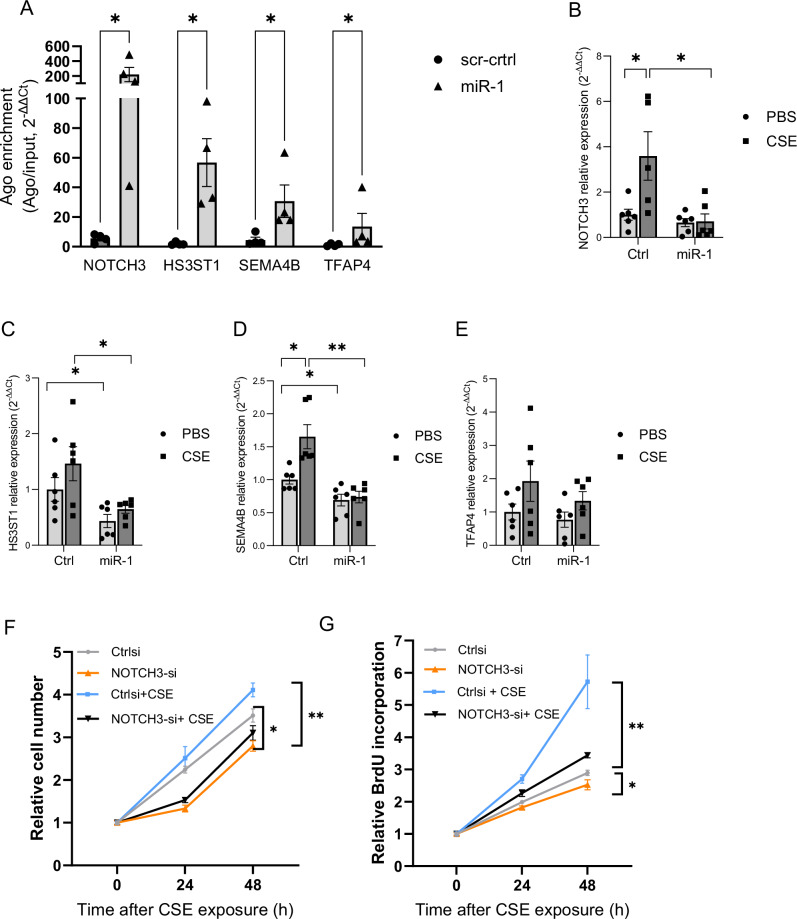
Cigarette smoke (CS) creates a "cancer field" in the lung that promotes malignant transformation. The molecular changes within this field are not fully characterized. We examined the significance of microRNA-1 (miR-1) downregulation as one of these changes. We found that tumor miR-1 levels in three non-small cell lung cancer cohorts show inverse correlations with the smoking burden. Lung MiR-1 levels follow a spatial gradient, have prognostic significance, and correlate inversely with the molecular markers of injury. In CS-exposed lungs, miR-1 is specifically downregulated in the endothelium. Exposure to CS induces angiogenesis by selectively degrading mature miR-1 via a vascular endothelial growth factor-driven pathway. Applying a multi-step molecular screen, we identified angiogenic genes regulated by miR-1 in the lungs of smokers. Knockdown of one of these genes, Notch homolog protein 3, simulates the anti-angiogenic effects of miR-1. These findings suggest that miR-1 can be used as an indicator of malignant transformation.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures




References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
