

Adaptive genomic signatures of globally invasive populations of the yellow fever mosquito Aedes aegypti
- PMID: 40155778
- PMCID: PMC11976285
- DOI: 10.1038/s41559-025-02643-5
Adaptive genomic signatures of globally invasive populations of the yellow fever mosquito Aedes aegypti
Abstract
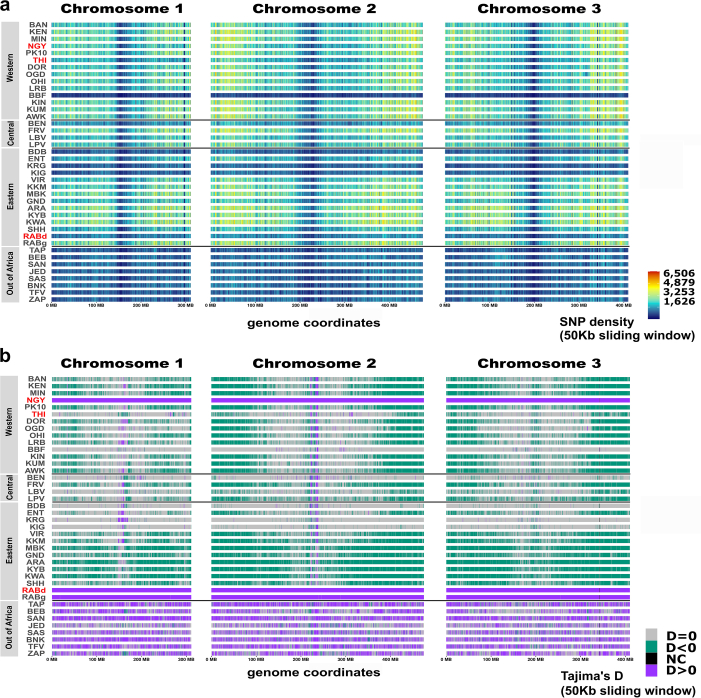
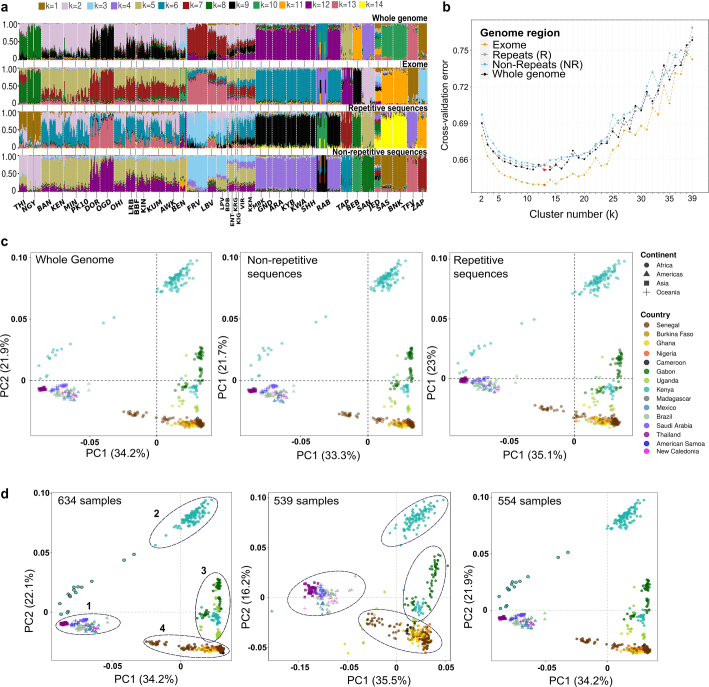
In the arboviral vector Aedes aegypti, adaptation to anthropogenic environments has led to a major evolutionary shift separating the domestic Aedes aegypti aegypti (Aaa) ecotype from the wild Aedes aegypti formosus (Aaf) ecotype. Aaa mosquitoes are distributed globally and have higher vectorial capacity than Aaf, which remained in Africa. Despite the evolutionary and epidemiological relevance of this separation, inconsistent morphological data and a complex population structure have hindered the identification of genomic signals distinguishing the two ecotypes. Here we assessed the correspondence between the geographic distribution, population structure and genome-wide selection of 511 Aaf and 123 Aaa specimens and report adaptive signals in 186 genes that we call Aaa molecular signatures. Our results indicate that Aaa molecular signatures arose from standing variation associated with extensive ancestral polymorphisms in Aaf populations and have been co-opted for self-domestication through genomic and functional redundancy and local adaptation. Overall, we show that the behavioural shift of Ae. aegypti mosquitoes to live in association with humans relied on the fine regulation of chemosensory, neuronal and metabolic functions, as seen in the domestication processes of rabbits and silkworms. Our results also provide a foundation for the investigation of new genic targets for the control of Ae. aegypti populations.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures












References
-
- Tchouassi, D. P., Agha, S. B., Villinger, J., Sang, R. & Torto, B. The distinctive bionomics of Aedes aegypti populations in Africa. Curr. Opin. Insect Sci.54, 100986 (2022). - PubMed
-
- Aubry, F. et al. Enhanced Zika virus susceptibility of globally invasive Aedes aegypti populations. Science370, 991–996 (2020). - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
