

Copy number normalization distinguishes differential signals driven by copy number differences in ATAC-seq and ChIP-seq
- PMID: 40155863
- PMCID: PMC11951689
- DOI: 10.1186/s12864-025-11442-y
Copy number normalization distinguishes differential signals driven by copy number differences in ATAC-seq and ChIP-seq
Abstract
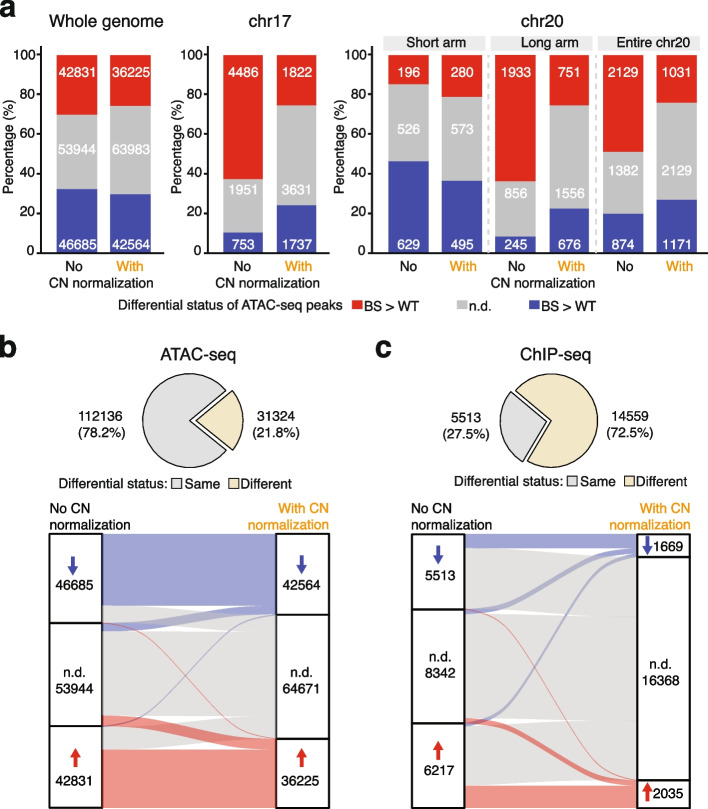
A common objective across ATAC-seq and ChIP-seq analyses is to identify differential signals across contrasted conditions. However, in differential analyses, the impact of copy number variation is often overlooked. Here, we demonstrated copy number differences among samples could drive, if not dominate, differential signals. To address this, we propose a pipeline featuring copy number normalization. By comparing the averaged signal per gene copy, it effectively segregates differential signals driven by copy number from other factors. Further applying it to Down syndrome unveiled distinct dosage-dependent and -independent changes on chromosome 21. Thus, we recommend copy number normalization as a general approach.
Keywords: ATAC-seq; Aneuploidy; ChIP-seq; Copy number normalization; Copy number variation; Differential analysis; Dosage effects; Down syndrome.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Competing interests: The authors declare no competing interests.
Figures




References
-
- Klemm SL, Shipony Z, Greenleaf WJ. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet. 2019;20:207–20. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
