

A one-base therapeutic insertion in the HBG2 distal promoter reactivates γ-globin expression
- PMID: 40156013
- PMCID: PMC11951516
- DOI: 10.1186/s40164-025-00626-7
A one-base therapeutic insertion in the HBG2 distal promoter reactivates γ-globin expression
Abstract
Background: The reactivation of developmental silenced γ-globin genes (HBG1/2) has shown promise as a therapeutic strategy for improving symptoms of β-hemoglobinopathies. Currently, the focus of therapeutic targets is primarily on the major fetal hemoglobin suppressors, such as BCL11A and ZBTB7A and of their binding sites on the proximal HBG promoter. However, the role of the distal HBG promoter in regulating gene expression remains to be explored.
Methods: We used CRISPR/Cas9 system to edit the distal HBG promoter. In vitro and in vivo assays, as well as engrafted NCG-Kit-V831M mice, were used for functional validation and mechanistic studies.
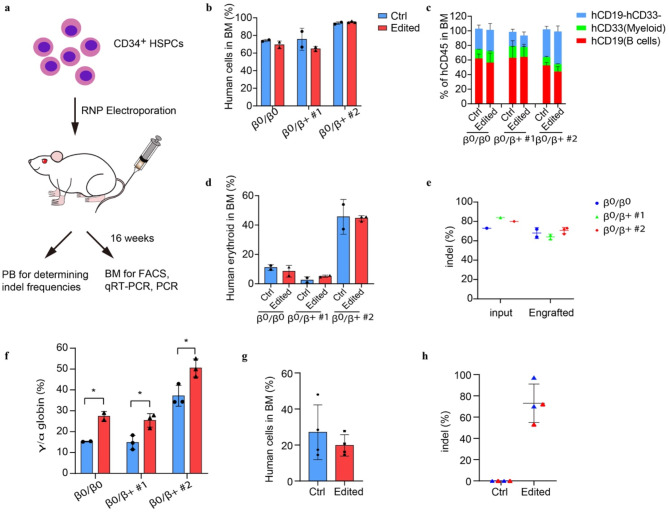
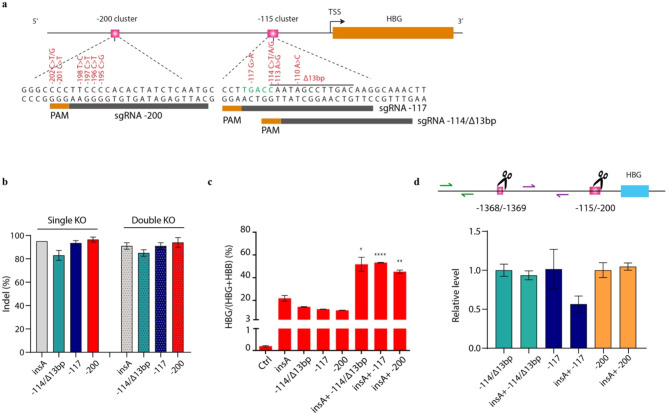
Results: We discovered an insertion of nucleotide A (insA) between - 1368 and - 1369 bp upstream of the TSS in HBG2 resulting in remarkable increase in γ-globin expression in HUDEP-2 cells. We also observed elevated γ-globin expression in human CD34+ erythroid progenitor cells from healthy individuals and those with β-thalassemia when introducing insA mutation. Similarly, engrafted NCG-Kit-V831M mice showed increased γ-globin expression. Importantly, neither did insA have any off-target effects nor did it affect the maturation of erythroid cells. Furthermore, we found that the insA mutation created a binding site for the transcription activator FOXO3, which was activated by AMPK. Additionally, introducing insA specifically demethylated the - 162 CpG site on HBG promoter by reducing the enrichment of DNA methyltransferase 3 A (DNMT3A). At the same time, it activated histone modifications and RNA polymerase II (Pol II) in both distal and proximal HBG promoter and might inhibit the binding of BCL11A and ZBTB7A on -115 and - 200 sites on the HBG promoter respectively. In addition, combination of insA and the - 115 or -200 editing targets resulted in an amplify effect in reactivating γ-globin genes expression.
Conclusions: Overall, we presented the preclinical data to support the role of insA on regulating γ-globin expression using CD34+ HSPC cells derived from healthy donors or patients with β-thalassemia, and subsequently engrafted mice. Our study suggests that introducing insA mutation leads to significantly boosted fetal globin levels and uncovers new safe therapeutic target or strategy for β-hemoglobinopathies.
Keywords: HBG2 distal promoter; CD34+ HSPC cells; FOXO3; Methylation; One-base therapeutic insertion; Therapeutic target; β-thalassemia.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: The healthy donors and patients with β-thalassemia were enrolled in Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong, China. Informed consents were obtained and the performants were complying with relevant ethnic regulations. β-thalassemia patient CD34+ HSPCs were isolated from plerixafor mobilized peripheral blood following Guangdong Women and Children Hospital institutional review board approval and patient informed consent. All animal experiments were approved by the Guangdong Women and Children Hospital Institutional Animal Care and Use Committee. Competing interests: The authors declare no competing interests.
Figures





Similar articles
-
Epigenetic inactivation of ERF reactivates γ-globin expression in β-thalassemia.Am J Hum Genet. 2021 Apr 1;108(4):709-721. doi: 10.1016/j.ajhg.2021.03.005. Epub 2021 Mar 17. Am J Hum Genet. 2021. PMID: 33735615 Free PMC article.
-
Disrupting ZBTB7A or BCL11A binding sites reactivates fetal hemoglobin in erythroblasts from healthy and β0-thalassemia/HbE individuals.Sci Rep. 2025 Jul 15;15(1):25580. doi: 10.1038/s41598-025-10791-8. Sci Rep. 2025. PMID: 40665149 Free PMC article.
-
CRISPR/Cas9-based multiplex genome editing of BCL11A and HBG efficiently induces fetal hemoglobin expression.Eur J Pharmacol. 2022 Mar 5;918:174788. doi: 10.1016/j.ejphar.2022.174788. Epub 2022 Jan 28. Eur J Pharmacol. 2022. PMID: 35093321
-
Precision Editing as a Therapeutic Approach for β-Hemoglobinopathies.Int J Mol Sci. 2023 May 31;24(11):9527. doi: 10.3390/ijms24119527. Int J Mol Sci. 2023. PMID: 37298481 Free PMC article. Review.
-
Wake-up Sleepy Gene: Reactivating Fetal Globin for β-Hemoglobinopathies.Trends Genet. 2018 Dec;34(12):927-940. doi: 10.1016/j.tig.2018.09.004. Epub 2018 Oct 1. Trends Genet. 2018. PMID: 30287096 Review.
References
-
- Mettananda S, Higgs DR. Molecular basis and genetic modifiers of thalassemia. Hematol Oncol Clin N. 2018;32:177–91. - PubMed
-
- Needs T, Gonzalez-Mosquera LF, Lynch DT. Beta thalassemia. StatPearls. Treasure Island (FL); 2020.
-
- Martyn GE, et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat Genet. 2018;50:498–503. - PubMed
Grants and funding
- U20A20353/National Natural Science Foundation of China
- 82100136/National Natural Science Foundation of China
- 0720240246/"Guangdong Special Support Plan" Provincial Health and Health Commission (Health Talent) Project
- 2023A1515010254/the Basic and Applied Basic Research Foundation of GuangDong Province
- 2025A03J4205/Guangzhou Municipal Science and Technology Project
LinkOut - more resources
Full Text Sources
Research Materials
