

Practical compendium of antiarrhythmic drugs: a clinical consensus statement of the European Heart Rhythm Association of the European Society of Cardiology
- PMID: 40159403
- PMCID: PMC12367031
- DOI: 10.1093/europace/euaf076
Practical compendium of antiarrhythmic drugs: a clinical consensus statement of the European Heart Rhythm Association of the European Society of Cardiology
Abstract
The European Heart Rhythm Association Practical Compendium of Anti-arrhythmic Drugs (AADs) offers advice on these drugs, focusing on their clinical use and the global impact of cardiac arrhythmias. This document aims to provide practical instructions to clinicians in arrhythmia management through pharmacological strategies. The compendium highlights persistent challenges in arrhythmia treatment, including clinical constraints, procedural risks, and the complexity of certain arrhythmias. Notably, atrial fibrillation is highly prevalent, and the demand for invasive treatment often surpasses the capacity of existing healthcare systems. As a result, pharmacological management remains essential. This is particularly relevant for patients with cardiac implantable electronic devices or channelopathies, where ablation is often not a suitable option. Anti-arrhythmic drugs play a pivotal role in these scenarios. The compendium introduces the ABC framework for AAD therapy: A (Appropriate therapy), for patients in whom AADs are the best therapeutic option; B (Backup therapy), as adjunctive treatment to invasive procedures, such as catheter ablation; and C (Complementary therapy), in combination with other therapies. The document provides detailed insights into the mechanisms of action, efficacy, safety profiles, and drug interactions of each class of AADs. Additionally, the compendium covers practical considerations, including initiation, combination strategies, monitoring, follow-up, special populations, and adverse effect management, with an emphasis on pro-arrhythmia risk mitigation. It also explores the integration of AADs with other therapeutic modalities, promoting a synergistic approach to optimize patient outcomes. In summary, this compendium serves as an indispensable resource for clinicians, offering practical advice and evidence-based insights to navigate the complexities of arrhythmia management effectively.
Keywords: Adverse drug reactions; Anti-arrhythmic drug combinations; Anti-arrhythmic drugs; Arrhythmia; Atrial fibrillation; Drug interactions; Mechanisms; Pharmacology; Ventricular arrhythmias.
© The European Society of Cardiology 2025.
Conflict of interest statement
Conflict of interest: A.G. has received funding from the EU Horizon 2020 programme: MAESTRIA Consortium grant number 952166 and speaker fees from Astra Zeneca, Boehringer Ingelheim, BMS/Pfizer, Daiichi Sankyo, and Medtronic. A.J.C. has received personal consulting fees from Acesion, InCarda, Menarini, Milestone, Sanofi, Anthos, Bayer, Daiichi Sankyo, Pfizer, Abbott, Biosense Webster, Biotronik, Boston Scientific, Medtronic, GlaxoSmithKline, and Johnson & Johnson. C.B.-L. has received fees and honoraria for lectures, education, and scientific advice from Abbott, Biosense Webster, Bayer, Sanofi, Organon, Philips, Medtronic, Boston Sci, and Cathprint. D.D. has received fees and honoraria for lectures and education from Daiichi Sankyo. G.B. reports small speaker fees from Bayer, Boehringer Ingelheim, Boston, Daiichi Sankyo, Janssen, and Sanofi outside of the submitted work. He is also the principal investigator of the ARISTOTELES (Applying ARtificial Intelligence to define clinical trajectorieS for personalized predicTiOn and early deTEction of comorbidity and muLtimorbidity pattErnS) project that received funding from the European Union within the Horizon 2020 research and innovation programme (grant no. 101080189). H.J.G.M.C. has received fees and honoraria for lectures, education, and scientific advice from InCarda Therapeutics, Roche, Sanofi, Atricure, Medtronic, and Armgo. J.L.M. has received fees and honoraria for lectures, education, and scientific advice from Abbott, Biosense Webster, Biotronik, iRhythm Technologies, MicroPort, and Zoll. He is also a member of the steering committe in the EHRA-PATHS (Addressing Multimorbidity in Elderly Atrial Fibrillation Patients Through Interdisciplinary, Tailored, Patient-Centered Care Pathways, GA 945260) and PROFID (Implementation of Personalized Risk Prediction and Prevention of Sudden Cardiac Death After Myocardial Infarction, GA 847999) projects, both funded by the European Union under the Horizon 2020 Research and Innovation Programme. J.A.R. reports being an investigator for Sanofi, InCarda Therapeutics, Johnson & Johnson, and Amarin and as a consultant for Sanofi and Acesion. J.T.-F. has received fees and honoraria for lectures, education, and scientific advice from Cytokinetics, Johnson and Johnson, MicroPort, Leo Pharma, and Boston Scientific. SHH has received fees and honoraria for lectures, education, and scientific advice from Sanofi, BI, Pfizer, BMS, Daiichi, and Incardia. The remaining authors declared no conflict of interest.
Figures
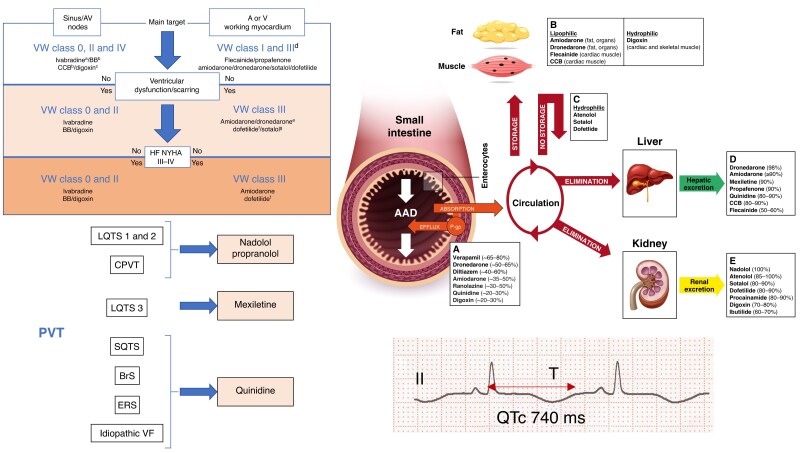
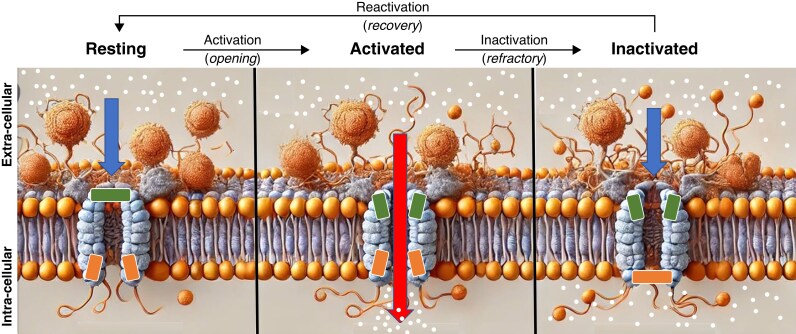




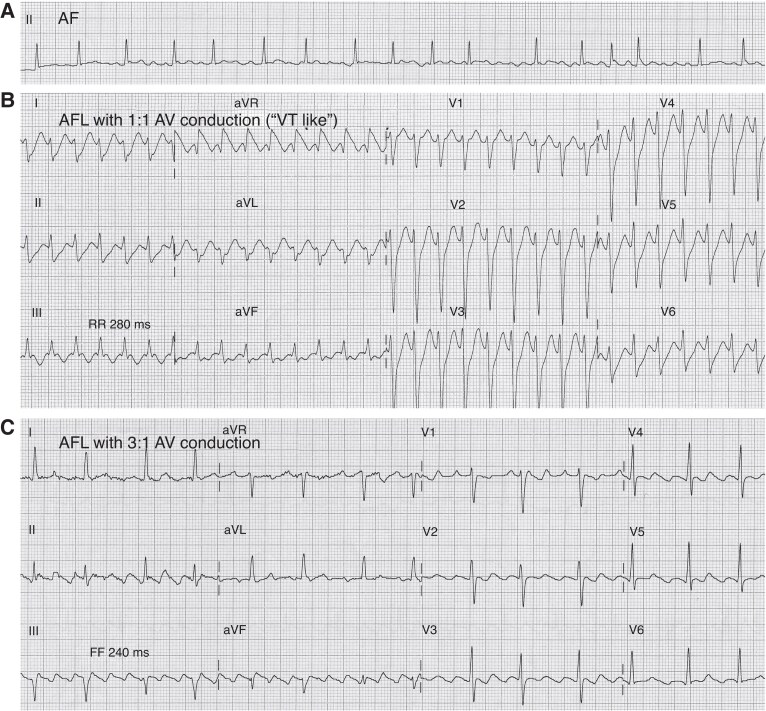
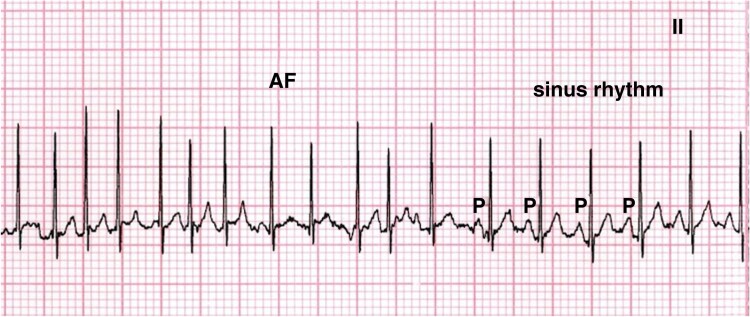
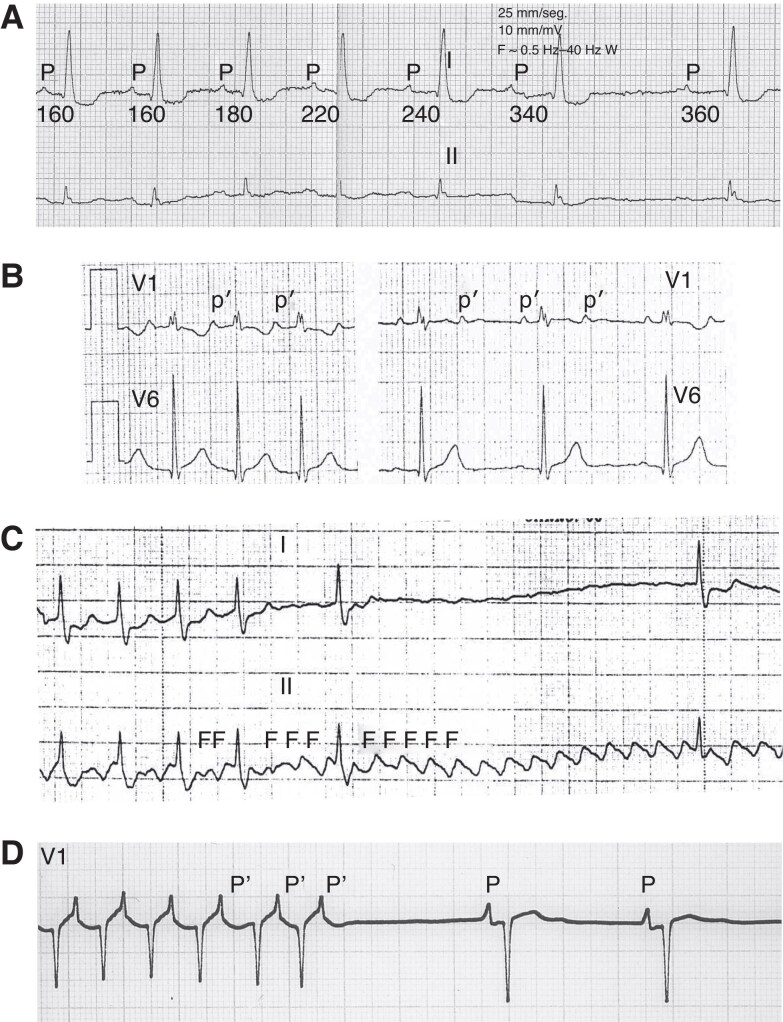
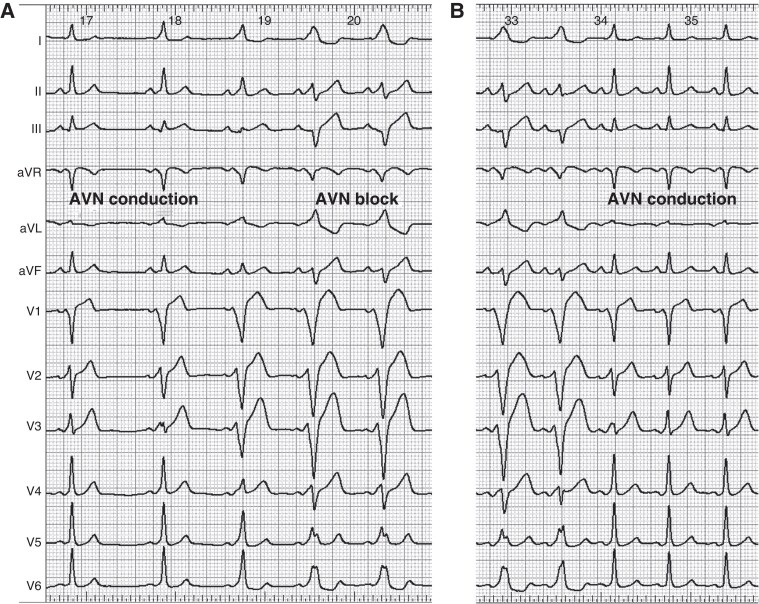
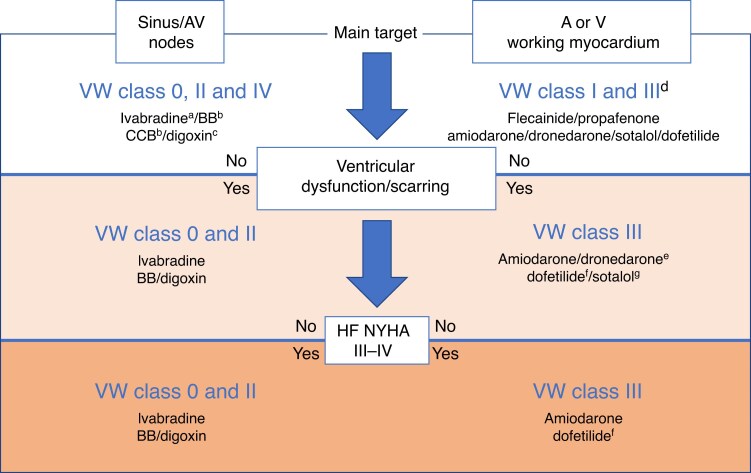
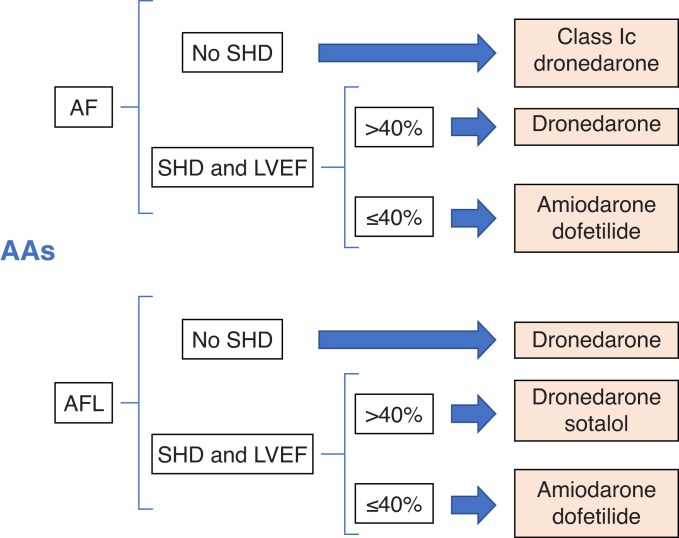

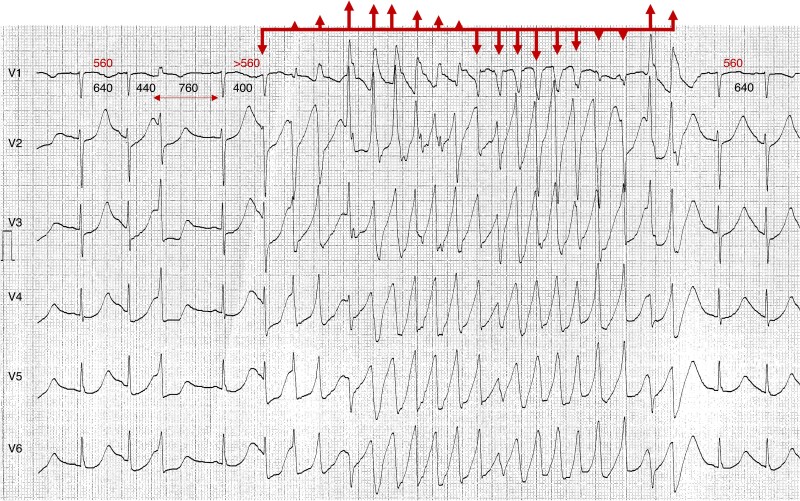
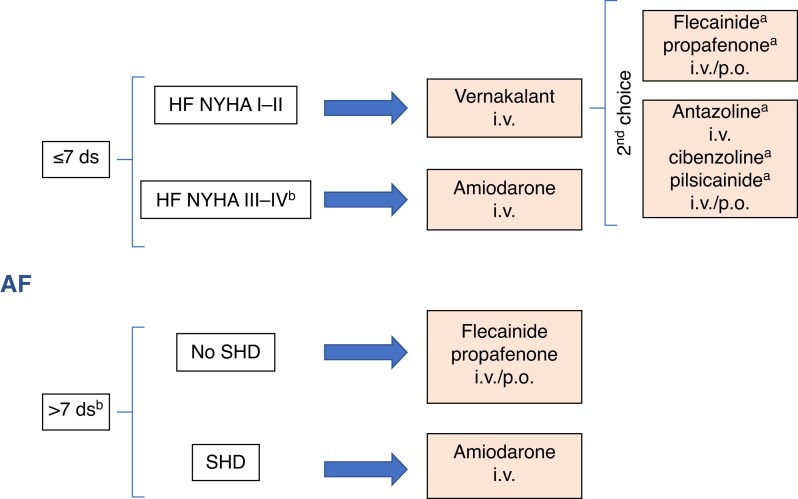
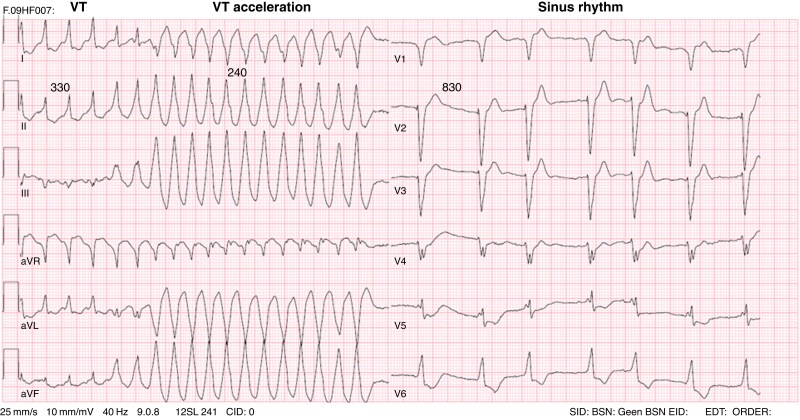
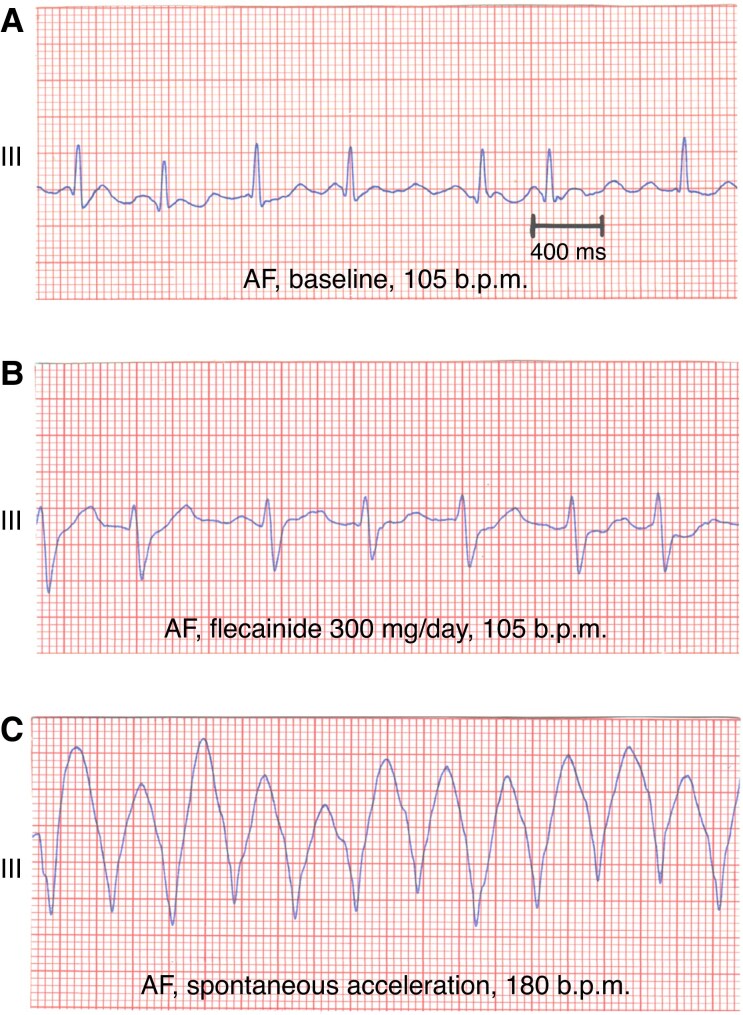



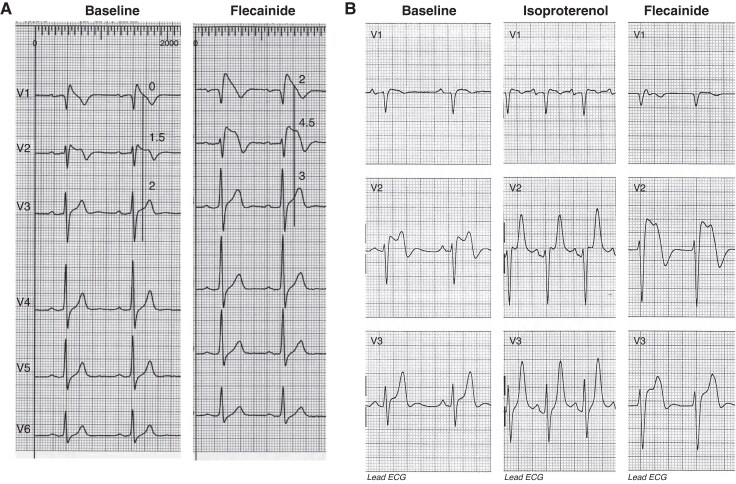
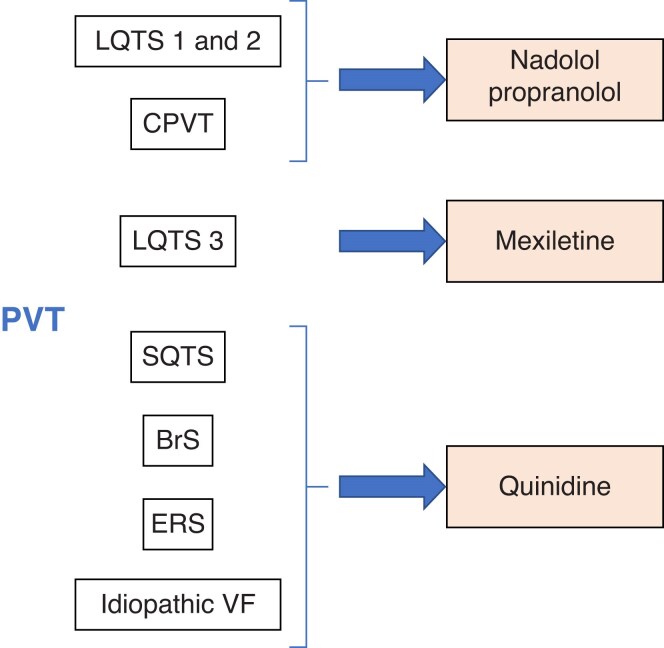
References
-
- Saksena S, Ken-Opurum J, McKindley DS, Preblick R, Rashkin J, Aldaas OM et al. Arrhythmia recurrence and rhythm control strategies after catheter ablation of newly diagnosed atrial fibrillation (ARRC-AF study). JACC Clin Electrophysiol 2025:S2405-500X(24)01027-2. - PubMed
-
- Heijman J, Voigt N, Nattel S, Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res 2014;114:1483–99. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
