

Gut metagenomes reveal interactions between dietary restriction, ageing and the microbiome in genetically diverse mice
- PMID: 40164832
- PMCID: PMC12360852
- DOI: 10.1038/s41564-025-01963-3
Gut metagenomes reveal interactions between dietary restriction, ageing and the microbiome in genetically diverse mice
Abstract
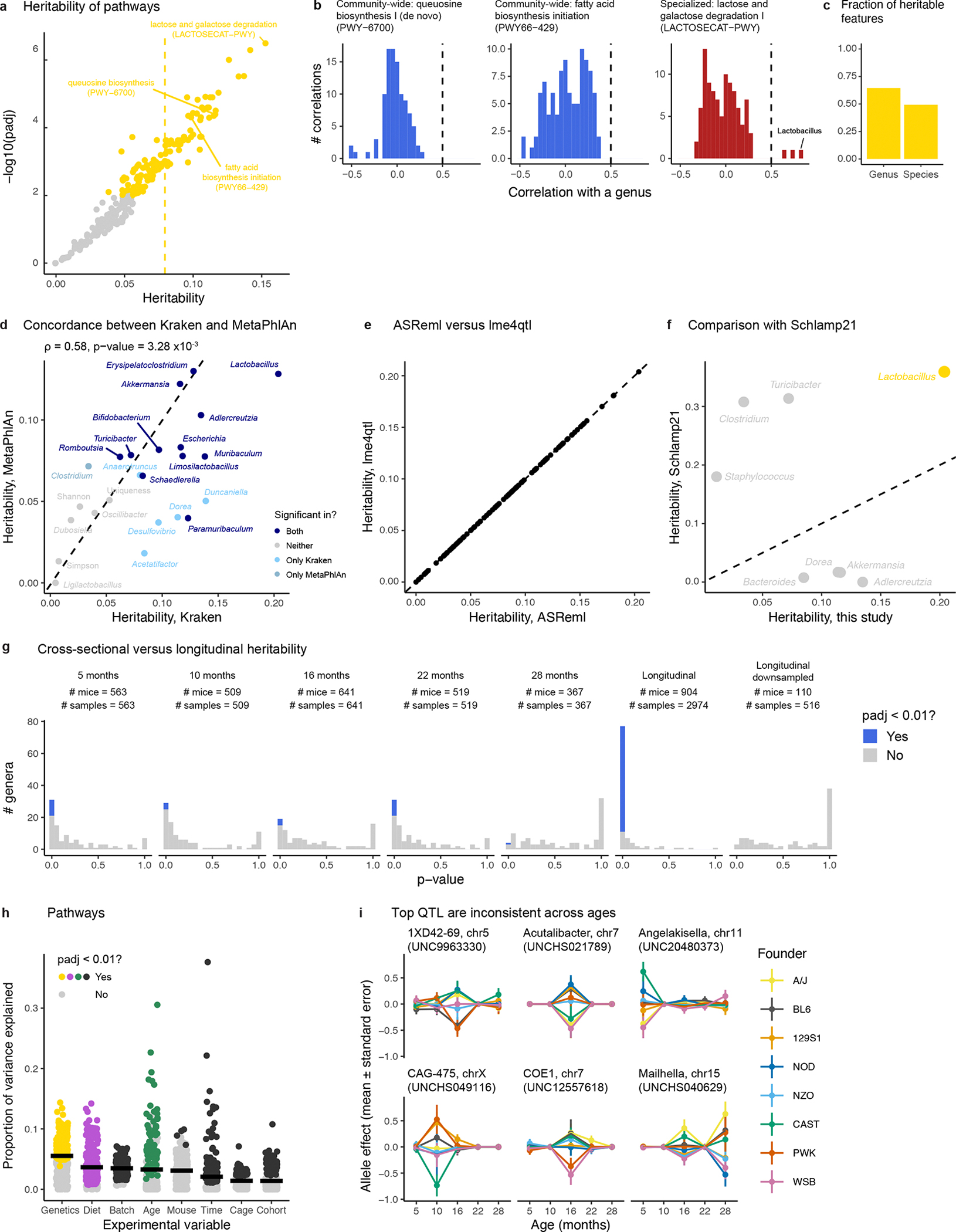
The gut microbiome changes with age and has been proposed to mediate the benefit of lifespan-extending interventions such as dietary restriction. However, the causes and consequences of microbiome ageing and the potential of such interventions remain unclear. Here we analysed 2,997 metagenomes collected longitudinally from 913 deeply phenotyped, genetically diverse mice to investigate interactions between the microbiome, ageing, dietary restriction (caloric restriction and fasting), host genetics and a range of health parameters. Among the numerous age-associated microbiome changes that we find in this cohort, increased microbiome uniqueness is the most consistent parameter across a second longitudinal mouse experiment that we performed on inbred mice and a compendium of 4,101 human metagenomes. Furthermore, cohousing experiments show that age-associated microbiome changes may be caused by an accumulation of stochastic environmental exposures (neutral theory) rather than by the influence of an ageing host (selection theory). Unexpectedly, the majority of taxonomic and functional microbiome features show small but significant heritability, and the amount of variation explained by host genetics is similar to ageing and dietary restriction. We also find that more intense dietary interventions lead to larger microbiome changes and that dietary restriction does not rejuvenate the microbiome. Lastly, we find that the microbiome is associated with multiple health parameters, including body composition, immune components and frailty, but not lifespan. Overall, this study sheds light on the factors influencing microbiome ageing and aspects of host physiology modulated by the microbiome.
© 2025. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing interests: K.M.W., A.R., F.H., Z.C., G.V.P., M.M., R.L.C., D.B., A.F. and A.D.F. are current or former employees of Calico Life Sciences LLC. The other authors declare no competing interests.
Figures















References
MeSH terms
Grants and funding
- DP1 DK140021/DK/NIDDK NIH HHS/United States
- R01 HL150359/HL/NHLBI NIH HHS/United States
- R01 DK129691/DK/NIDDK NIH HHS/United States
- R01 EY031209/EY/NEI NIH HHS/United States
- T32 HG000046/HG/NHGRI NIH HHS/United States
- R01 EY030192/EY/NEI NIH HHS/United States
- DP2 AG067492/AG/NIA NIH HHS/United States
- R01 NS134976/NS/NINDS NIH HHS/United States
- P30 AG038070/AG/NIA NIH HHS/United States
- F30 AG080958/AG/NIA NIH HHS/United States
- R01 GM125301/GM/NIGMS NIH HHS/United States
- R35 GM146980/GM/NIGMS NIH HHS/United States
- R01 HL113147/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
