

Genetic editing of primary human dorsal root ganglion neurons using CRISPR-Cas9
- PMID: 40169710
- PMCID: PMC11961745
- DOI: 10.1038/s41598-025-91153-2
Genetic editing of primary human dorsal root ganglion neurons using CRISPR-Cas9
Abstract
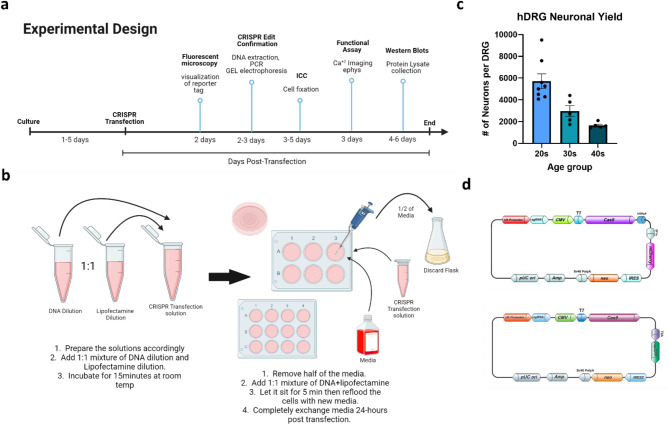
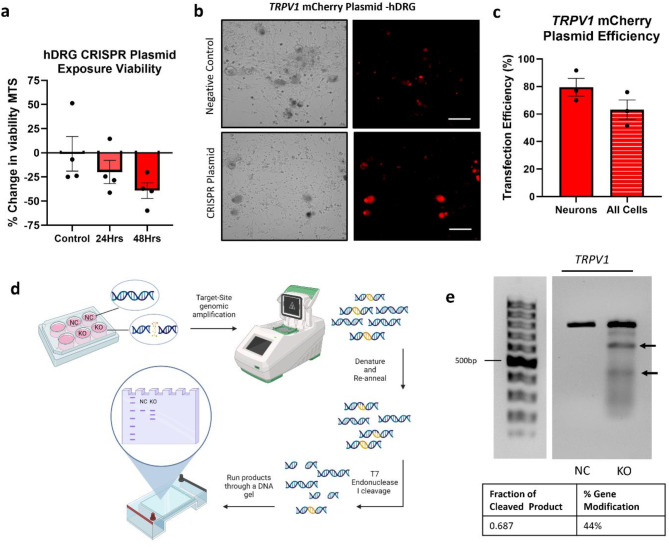
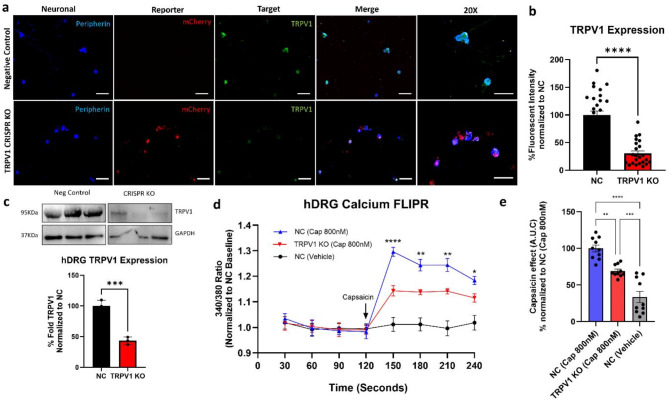
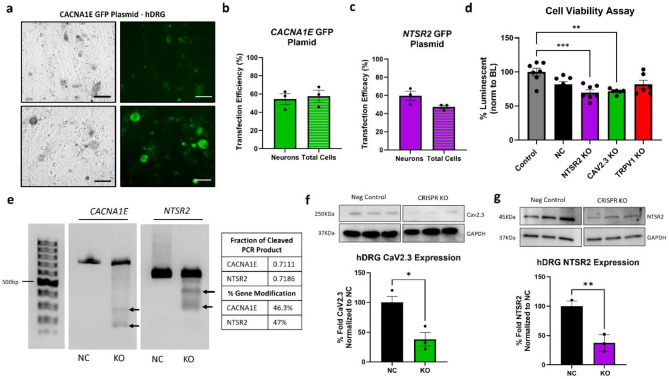
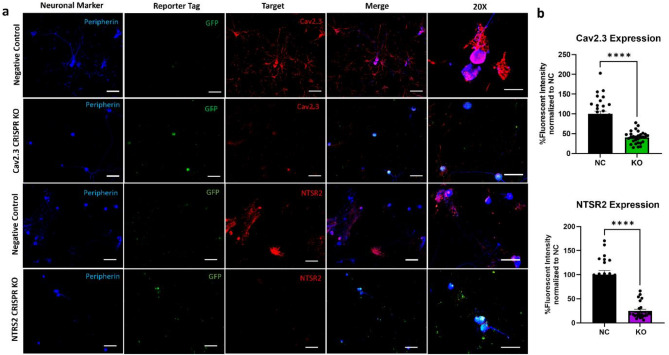
CRISPR-Cas9 is now the leading method for genome editing and is advancing for the treatment of human disease. CRIPSR has promise in treating neurological diseases, but traditional viral-vector-delivery approaches have neurotoxicity limiting their use. Here we describe a simple method for non-viral transfection of primary human DRG (hDRG) neurons for CRISPR-Cas9 editing. We edited TRPV1, NTSR2, and CACNA1E using a lipofection method with CRISPR-Cas9 plasmids containing reporter tags (GFP or mCherry). Transfection was successfully demonstrated by the expression of the reporters two days post-administration. CRISPR-Cas9 editing was confirmed at the genome level with a T7-endonuclease-I assay; protein level with immunocytochemistry and Western blot; and functional level through capsaicin-induced Ca2+ accumulation in a high-throughput compatible fluorescent imaging plate reader (FLIPR) system. This work establishes a reliable, target specific, non-viral CRISPR-Cas9-mediated genetic editing in primary human neurons with potential for future clinical application for sensory diseases.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures
References
-
- Doudna, J. A. & Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science346, 1258096 (2014). - PubMed
-
- Jiang, F. & Doudna, J. A. CRISPR-Cas9 structures and mechanisms. Annu. Rev. Biophys.46, 505–529 (2017). - PubMed
-
- Hsu, S., Huang, G. S., Ho, T. T. & Feng, F. Efficient gene Silencing in mesenchymal stem cells by substrate-mediated RNA interference. Tissue Eng. Part. C: Methods. 20, 916–930 (2014). - PubMed
-
- Barrangou, R. & Doudna, J. A. Applications of CRISPR technologies in research and beyond. Nat. Biotechnol.34, 933–941 (2016). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
