

Reassessment of FBN1 variants of uncertain significance using updated ClinGen guidance for PP1/BS4 and PP4 criteria
- PMID: 40169830
- PMCID: PMC12048594
- DOI: 10.1038/s41431-025-01826-9
Reassessment of FBN1 variants of uncertain significance using updated ClinGen guidance for PP1/BS4 and PP4 criteria
Abstract
Marfan syndrome (MFS) is a genetic disorder caused by an FBN1 variant and is diagnosed based on the revised Ghent criteria, which incorporate clinical manifestations and genetic testing. Up-to-date FBN1 variant interpretation is crucial for proper diagnosis and management of MFS; however, some FBN1 variants of uncertain significance (VUSs) remain inconclusive despite applying Clinical Genome Resource (ClinGen) FBN1-specific guideline. Recently, the ClinGen guidance for PP1/BS4 co-segregation and PP4 phenotype specificity criteria (new PP1/PP4 criteria) were released. Here, we performed reassessment of FBN1 VUSs using these new PP1/PP4 criteria. FBN1 VUSs collected from December 2015 to April 2024 were reassessed according to the ClinGen FBN1-specific guideline and new PP1/PP4 criteria. Medical records and previous studies were reviewed to evaluate the phenotype-specificity of evidence based on the revised Ghent criteria. Collectively, 927 patients with suspected MFS underwent FBN1 sequencing and 72 VUSs were detected. When applying the FBN1-specific guideline only, of 72 VUSs, 29 (40.3%) were reclassified as pathogenic variants (PVs) or likely PVs (LPVs). When additionally applying the new PP1/PP4 criteria, 16 (37.2%) of the remaining 43 VUSs were reclassified as LPVs. After reassessing FBN1 VUSs according to the new PP1/PP4 criteria, the rate of reclassification from VUS to PV/LPV significantly increased from 40.3% to 62.5%. The new PP1/PP4 criteria provide sufficient evidence for evaluating the pathogenicity of FBN1 variants detected in MFS patients fulfilling the revised Ghent criteria and will be helpful in clinical analysis.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests. Ethical approval: The study was approved by the IRB of SMC, Seoul, Korea (approval no. 2024-02-006). The requirement of informed consent was waived due to its retrospective nature.
Figures
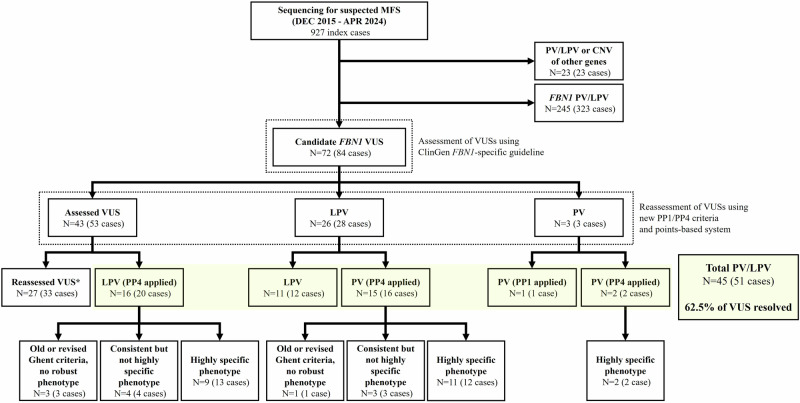
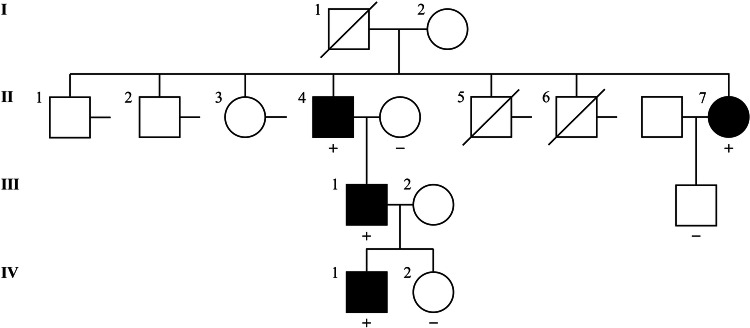
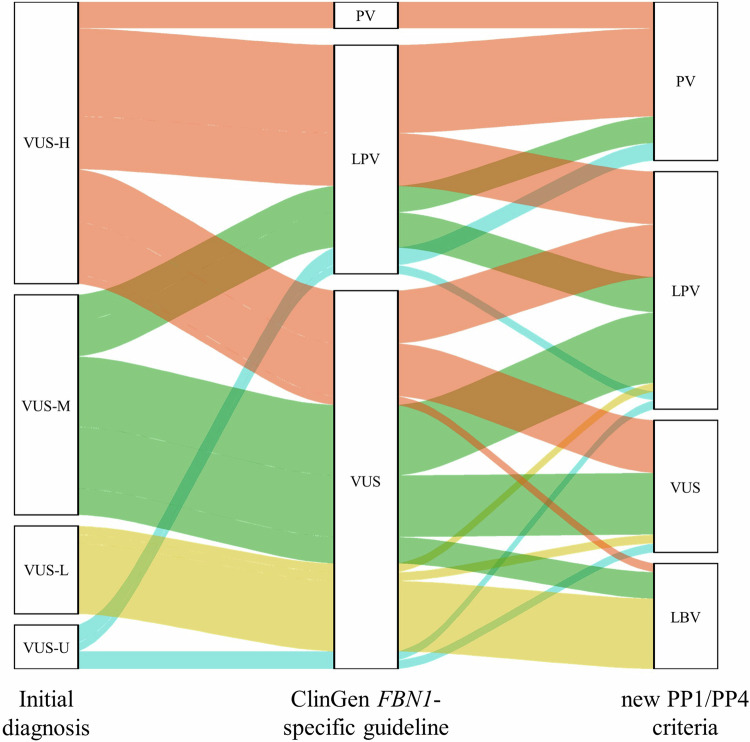
References
-
- Meester JAN, Peeters S, Van Den Heuvel L, Vandeweyer G, Fransen E, Cappella E, et al. Molecular characterization and investigation of the role of genetic variation in phenotypic variability and response to treatment in a large pediatric Marfan syndrome cohort. Genet Med. 2022;24:1045–53. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
