

ATM-dependent DNA damage response constrains cell growth and drives clonal hematopoiesis in telomere biology disorders
- PMID: 40179146
- PMCID: PMC11996883
- DOI: 10.1172/JCI181659
ATM-dependent DNA damage response constrains cell growth and drives clonal hematopoiesis in telomere biology disorders
Abstract
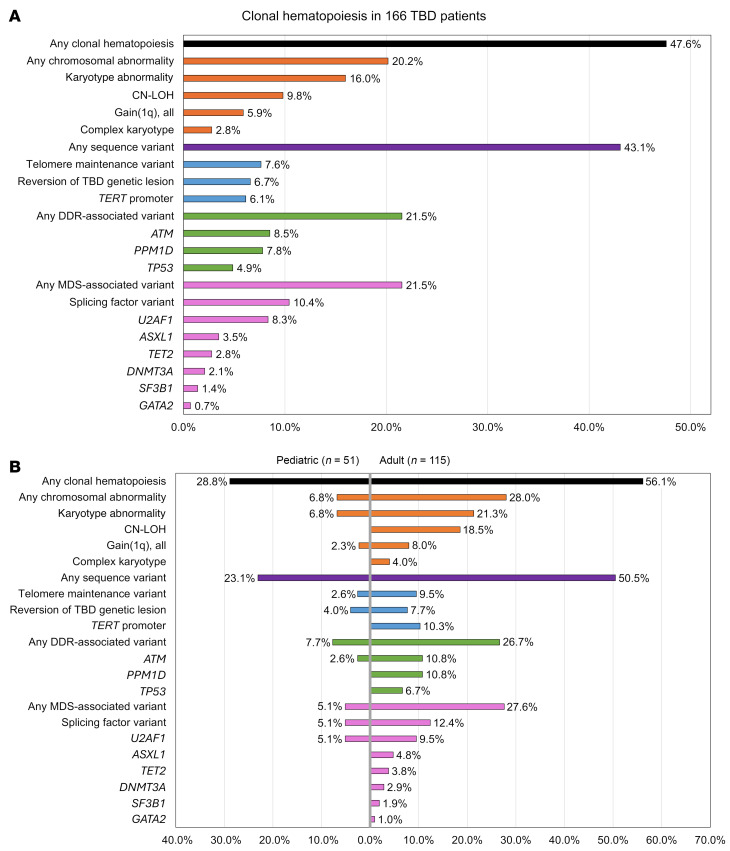
Telomere biology disorders (TBDs) are genetic diseases caused by defective telomere maintenance. TBD patients often develop bone marrow failure and have an increased risk of myeloid neoplasms. To better understand the factors underlying hematopoietic outcomes in TBD, we comprehensively evaluated acquired genetic alterations in hematopoietic cells from 166 pediatric and adult TBD patients. Of these patients, 47.6% (28.8% of children, 56.1% of adults) had clonal hematopoiesis. Recurrent somatic alterations involved telomere maintenance genes (7.6%), spliceosome genes (10.4%, mainly U2AF1 p.S34), and chromosomal alterations (20.2%), including 1q gain (5.9%). Somatic variants affecting the DNA damage response (DDR) were identified in 21.5% of patients, including 20 presumed loss-of-function variants in ataxia-telangiectasia mutated (ATM). Using multimodal approaches, including single-cell sequencing, assays of ATM activation, telomere dysfunction-induced foci analysis, and cell-growth assays, we demonstrate telomere dysfunction-induced activation of the ATM-dependent DDR pathway with increased senescence and apoptosis in TBD patient cells. Pharmacologic ATM inhibition, modeling the effects of somatic ATM variants, selectively improved TBD cell fitness by allowing cells to bypass DDR-mediated senescence without detectably inducing chromosomal instability. Our results indicate that ATM-dependent DDR induced by telomere dysfunction is a key contributor to TBD pathogenesis and suggest dampening hyperactive ATM-dependent DDR as a potential therapeutic intervention.
Keywords: Clonal selection; Hematology; Hematopoietic stem cells; Oncology; Telomeres.
Conflict of interest statement
Figures













References
-
- Savage SA, Niewisch MR. Duskeratosis congenita and related telomere biology disorders. In: Gene Reviews. University of Washington; 2023:NBK22301.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
