

Decreased mitochondrial NAD+ in WRN deficient cells links to dysfunctional proliferation
- PMID: 40179319
- PMCID: PMC12074813
- DOI: 10.18632/aging.206236
Decreased mitochondrial NAD+ in WRN deficient cells links to dysfunctional proliferation
Abstract
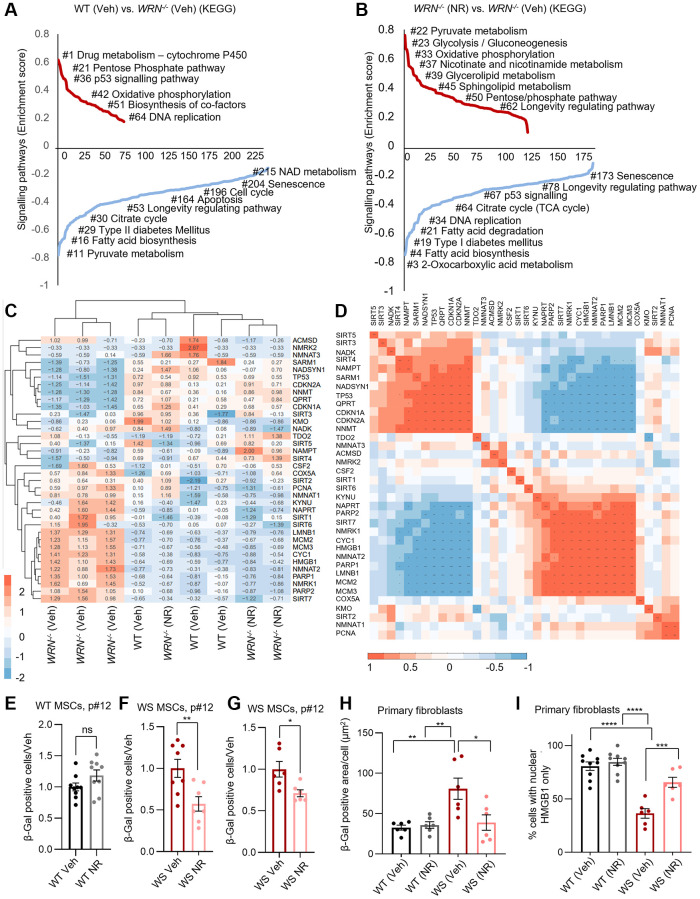
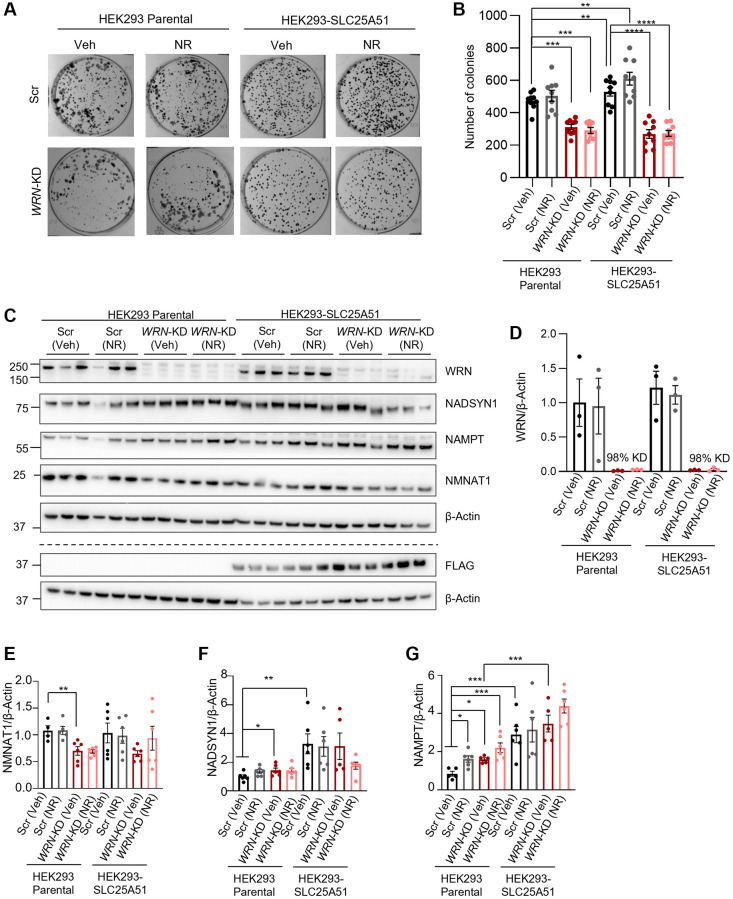
Werner syndrome (WS), caused by mutations in the RecQ helicase WERNER (WRN) gene, is a classical accelerated aging disease with patients suffering from several metabolic dysfunctions without a cure. While, as we previously reported, depleted NAD+ causes accumulation of damaged mitochondria, leading to compromised metabolism, how mitochondrial NAD+ changes in WS and the impact on WS pathologies were unknown. We show that loss of WRN increases senescence in mesenchymal stem cells (MSCs) likely related to dysregulation of metabolic and aging pathways. In line with this, NAD+ augmentation, via supplementation with nicotinamide riboside, reduces senescence and improves mitochondrial metabolic profiles in MSCs with WRN knockout (WRN-/-) and in primary fibroblasts derived from WS patients compared to controls. Moreover, WRN deficiency results in decreased mitochondrial NAD+ (measured indirectly via mitochondrially-expressed PARP activity), and altered expression of key salvage pathway enzymes, including NMNAT1 and NAMPT; ChIP-seq data analysis unveils a potential co-regulatory axis between WRN and the NMNATs, likely important for chromatin stability and DNA metabolism. However, restoration of mitochondrial or cellular NAD+ is not sufficient to reinstall cellular proliferation in immortalized cells with siRNA-mediated knockdown of WRN, highlighting an indispensable role of WRN in proliferation even in an NAD+ affluent environment. Further cell and animal studies are needed to deepen our understanding of the underlying mechanisms, facilitating related drug development.
Keywords: NAD+; Werner syndrome; mitochondria; premature aging; proliferation.
Conflict of interest statement
Figures


References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
