

Therapeutic potential of allosteric HECT E3 ligase inhibition
- PMID: 40179885
- PMCID: PMC12087876
- DOI: 10.1016/j.cell.2025.03.001
Therapeutic potential of allosteric HECT E3 ligase inhibition
Abstract
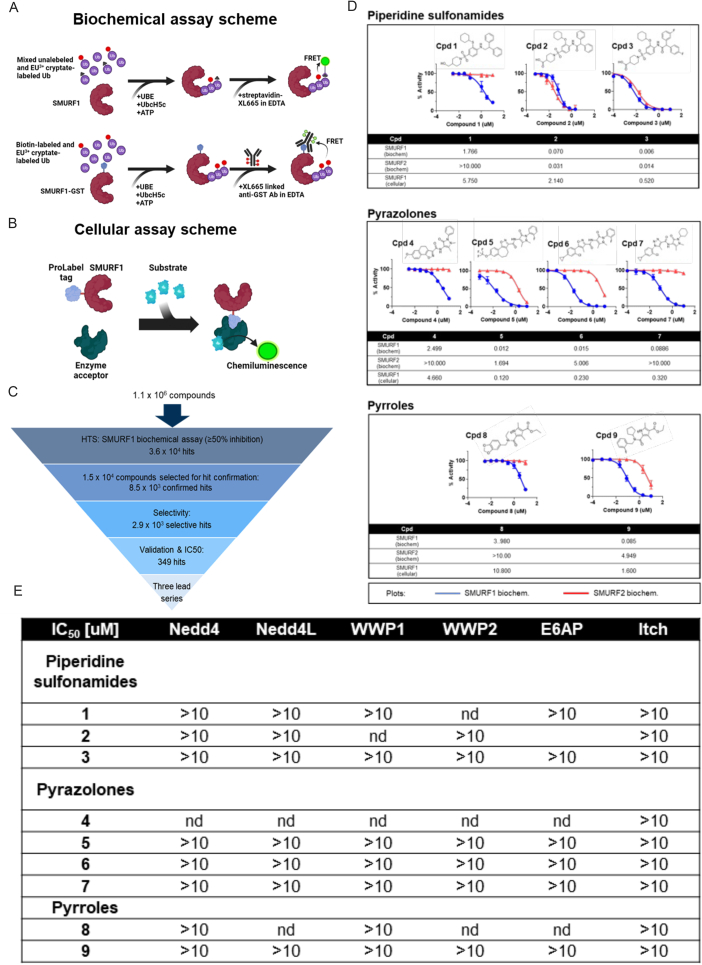
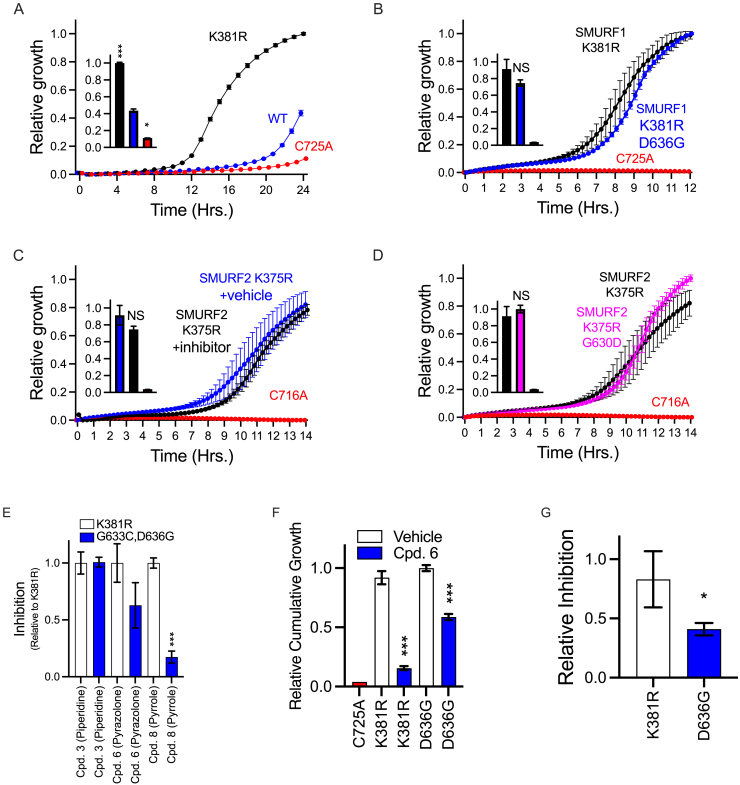
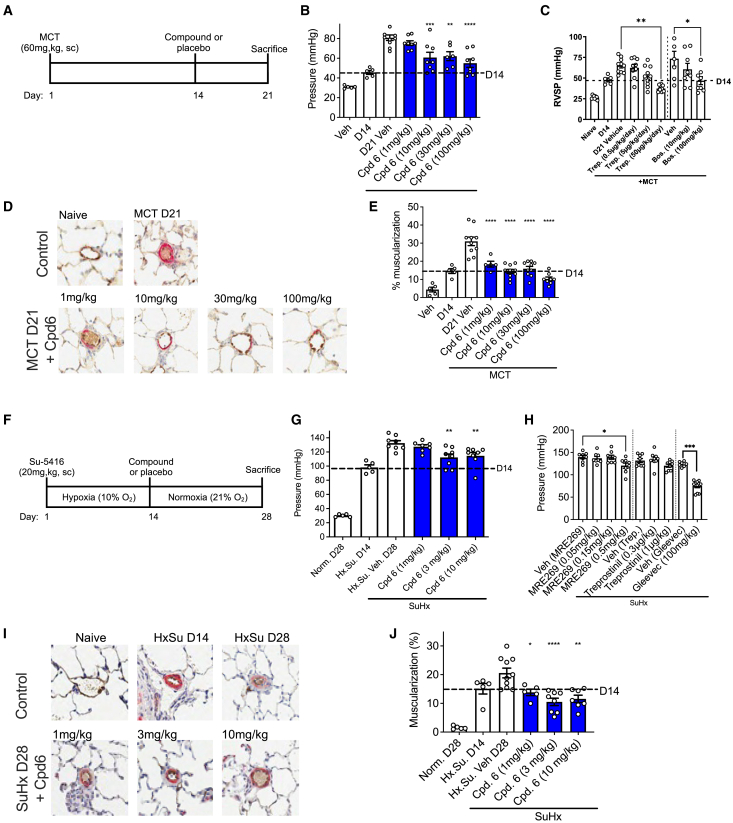
Targeting ubiquitin E3 ligases is therapeutically attractive; however, the absence of an active-site pocket impedes computational approaches for identifying inhibitors. In a large, unbiased biochemical screen, we discover inhibitors that bind a cryptic cavity distant from the catalytic cysteine of the homologous to E6-associated protein C terminus domain (HECT) E3 ligase, SMAD ubiquitin regulatory factor 1 (SMURF1). Structural and biochemical analyses and engineered escape mutants revealed that these inhibitors restrict an essential catalytic motion by extending an α helix over a conserved glycine hinge. SMURF1 levels are increased in pulmonary arterial hypertension (PAH), a disease caused by mutation of bone morphogenetic protein receptor-2 (BMPR2). We demonstrated that SMURF1 inhibition prevented BMPR2 ubiquitylation, normalized bone morphogenetic protein (BMP) signaling, restored pulmonary vascular cell homeostasis, and reversed pathology in established experimental PAH. Leveraging this deep mechanistic understanding, we undertook an in silico machine-learning-based screen to identify inhibitors of the prototypic HECT E6AP and confirmed glycine-hinge-dependent allosteric activity in vitro. Inhibiting HECTs and other glycine-hinge proteins opens a new druggable space.
Keywords: E6AP; HECT; SMURF1; allosteric inhibition; drug discovery; glycine hinge; pulmonary arterial hypertension; small molecule; ubiquitin E3 ligase; vascular remodeling.
Copyright © 2025 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests D.J.R., S.G.-G., C.Q., O.B., R.H., A.H., T.R., M.T., R.J., D. Sutton, J.R., J.-H.Z., S.W., S.C., J.O., S.S., T.T., M.H., M.J.T., G.J., R.E.J.B., J.S.C., R.L., N.S., N.O., J.T., and D. Shaw are current or former Novartis employees. Two patents have been submitted related to this work: WO2020079687A1 (G.P.) and US11672782B2 (D. Shaw and N.S.).
Figures










References
-
- Yang Y., Kitagaki J., Dai R.-M., Tsai Y.C., Lorick K.L., Ludwig R.L., Pierre S.A., Jensen J.P., Davydov I.V., Oberoi P., et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007;67:9472–9481. doi: 10.1158/0008-5472.CAN-07-0568. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
